静电纺丝制备g-C3N4/C纳米纤维及其可见光降解性能
2016-12-20杨佳佳蒋克望林雪梅应宗荣张文文
杨佳佳 蒋克望 林雪梅 应宗荣 张文文
(南京理工大学化工学院,南京210094)
静电纺丝制备g-C3N4/C纳米纤维及其可见光降解性能
杨佳佳蒋克望林雪梅应宗荣*张文文
(南京理工大学化工学院,南京210094)
采用g-C3N4纳米片与聚丙烯腈进行静电复合纺丝,再经预氧化和碳化制得g-C3N4/C纳米纤维。利用傅立叶变换红外光谱仪(FTIR)、X射线衍射仪(XRD)、拉曼光谱(Raman)和扫描电子显微镜(SEM)对样品结构和形貌进行表征,通过紫外-可见漫反射光谱(UV-Vis DRS)分析可见光响应性。研究表明,复合纳米纤维对罗丹明B表现出较好的可见光降解活性,源于无定形相/石墨相混合结构的碳基体能够降低g-C3N4的光生电子-空穴对复合的几率。复合纳米纤维膜在光催化降解搅拌条件下始终能保持完整,经过多次回收和光催化实验,对罗丹明B的光降解率依然较高,表现出较优异的循环利用稳定性。
类石墨相氮化碳;碳纤维;静电纺丝;复合纳米纤维;可见光;光降解
自福州大学王心晨教授课题组提出类石墨相氮化碳(g-C3N4)可以作为一种不含金属的光催化剂用于分解水制氢[1]以来,这一新型的光催化剂引起了材料和催化领域科研人员的兴趣[2-4],例如Liu等[5-6]使用Ag3PO4和Cu2O,Bai等[7]使用Ag分别对g-C3N4纳米片进行复合改性,得到了有明显催化活性的复合光催化剂。g-C3N4具有层状结构,理论上具有很大的比表面积,但事实上通过简单煅烧制备的g-C3N4是体相的块状聚集体,实际比表面积小得多,严重制约其光催化效率的发挥[8-10]。通过化学氧化法[11]、超声剥离法[12]和热剥离法[13],能够很好地将g-C3N4纳米片层从块体上剥离下来,剥离所得的纳米片层能够保持体相材料完整的晶体结构和相同的化学组成,再加上纳米片层材料所表现出的小尺寸效应和表面效应,极大地提高了其光催化活性[14]。直接使用剥离得到的g-C3N4纳米片用于光催化,在光催化过程中难以持续地保持片层结构,容易发生团聚,比表面积逐渐减小,光催化活性降低,特别是g-C3N4纳米片呈粉末状,容易流失,回收再利用困难,循环利用稳定性差。
静电纺丝是一种制备连续纳米纤维简单且有效的方法[15]。通过静电纺丝工艺控制可以制得独特的结构形貌,通过共混及复合静电纺丝可以很方便地获得多种组分复合的多元纳米纤维,包括无机/无机、无机/有机、有机/有机等复合型纳米纤维,这使纳米纤维进行各种功能设计及用途设计非常方便。由于静电纺丝纤维具有纳米尺寸、容易获得独特结构形貌和各种复合结构,使其在纳米电子器件[16]、电池和电极材料[17-18]、催化剂[19]等众多领域有着重要的应用。通过静电纺丝制备的纳米碳纤维直径细且具有较高的导电性[20-21],在作为光催化剂载体时,有利于光催化剂光生电子-空穴的分离,且对纳米级光催化剂起固定作用,在使用过程中阻碍光催化剂团聚,再加上碳纤维本身具有优异的柔韧性[22-23],使得光催化剂可以反复利用,循环利用稳定性大大提高。
本文采用聚丙烯腈(PAN)与g-C3N4纳米片进行静电混合纺丝,然后预氧化和碳化,以实现g-C3N4纳米片在碳纤维上的负载。研究发现,g-C3N4/C复合纤维内部的碳基体呈无定形相/石墨相混合结构,能够大大降低纤维表面g-C3N4(S)产生的光生电子-空穴对复合的几率,使复合纤维对罗丹明B溶液具有较优异的可见光催化活性,复合纤维在光催化降解搅拌条件中不会脆化,回收再用方便,循环利用稳定性好。
1 实验部分
1.1样品制备
1.1.1g-C3N4纳米片的制备
按照文献[24]制备g-C3N4纳米片。将三聚氰胺粉体置于真空管式高温烧结炉中,在550℃温度下于氮气氛下煅烧4 h得到体相g-C3N4。研细后加入陶瓷坩埚中于550℃温度下再次煅烧2 h,得到g-C3N4薄层,记作g-C3N4(F)。将0.2 g的g-C3N4薄层分散到20 mL的N,N-二甲基甲酰胺溶液中,超声剥离6 h得到g-C3N4纳米片,记作g-C3N4(S)。
1.1.2g-C3N4/PAN纳米纤维膜的制备
将2.604 g聚丙烯腈(PAN)加入到上述g-C3N4(S) DMF悬浮液当中,30℃水浴条件下搅拌5 h至PAN完全溶解,继续超声2 h后得到g-C3N4(S)悬浮分散纺丝液。在室温下进行静电纺丝,以覆盖铜网的铁板作为接收装置,电纺条件为:电压U=19 kV,接收距离d=15 cm,纺丝流速v=0.5 mL·h-1。将接收到的电纺纤维膜于80℃烘箱中干燥12 h,即得到g-C3N4/PAN纳米纤维膜。
1.1.3g-C3N4/C纳米纤维膜的制备
将上述g-C3N4/PAN纳米纤维膜裁剪成2 cm×3 cm矩形状,放入耐高温的陶瓷方舟中,置于真空管式高温烧结炉内。在空气气氛下以3℃·min-1速率从室温升温到290℃预氧化0.5 h;然后将管式炉抽至真空,通入氮气,控制合适的氮气流速,以6℃·min-1升温到600℃碳化0.5 h;最后,自然冷却至室温,得到g-C3N4/C纳米纤维。
1.2样品表征
采用傅立叶变换红外光谱仪(8400S,Shimadzu)测试FTIR光谱(FTIR),KBr压片法,扫描波数范围400~4 000 cm-1,分辨率8.0 cm-1,快速扫描每秒20次。采用X射线衍射仪(D8 Advance,Bruker AXS GmbH)测试XRD图,Cu Kα,波长λ=0.154 056 nm,工作电压40 kV,电流40 mA,扫描速度6°·min-1,扫描范围5°~80°。采用激光共焦显微拉曼光谱仪(in Via,Renishaw)测试拉曼光谱。采用紫外-可见漫反射光谱仪(UV-2500,Shimadzu)测试UV-Vis DRS光谱,测试波长200~800 nm,标准BaSO4参比物质,干法压片制样。采用扫描电子显微镜(FEI,FEG250, Quanta)表征微观结构和形貌,样品经表面喷金处理,加速电压15.0 kV。
1.3光催化降解实验
光催化实验在光化学反应仪(XPA系列,南京胥江机电厂)中进行。采用500 W的氙灯作为光源,使用滤光片滤掉420 nm以下波长的光,以30 mg活性物质的g-C3N4(F)、g-C3N4(S)和g-C3N4(S)/C纳米纤维膜为光催化剂,以50 mL的10 mg·L-1RhB溶液为目标降解物。先在暗光环境下搅拌吸附1 h以达到吸附-脱附平衡,开灯后每隔0.5 h取样一次,每次取样3 mL,光降解反应总时间3 h。取出来的样用高速离心机离心,去除试样中残留的催化剂,使用紫外-可见分光光度计检测其上层清液的吸光度值,根据公式W=(C0-C)/C×100%=(A0-A)/A0× 100%计算RhB的降解率W,式中C0和C、A0和A分别指开始光降解时RhB溶液和各取样RhB溶液中RhB的浓度、溶液的吸光度值。
采用g-C3N4(F)、g-C3N4(S)、g-C3N4/C纳米纤维膜在同一条件下5次循环降解目标污染物(RhB)实验用来评价光催化剂的催化稳定性。每次使用完后,离心取出样品,用去离子水洗涤3次,最后用乙醇洗,放入80℃烘箱烘干4 h,然后继续下一次光催化降解实验测试。
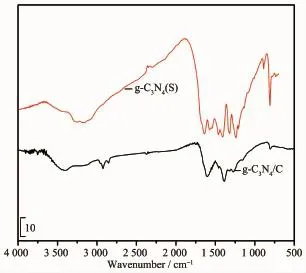
图1 g-C3N4(S)和g-C3N4/C纳米纤维膜的FTIR图Fig.1 FTIR spectra of g-C3N4(S)and g-C3N4/C nanofibers
2 结果与讨论
2.1FTIR分析
图1为g-C3N4(S)和g-C3N4/C纳米纤维膜的FTIR图。801 cm-1处为g-C3N4的3-s三嗪结构特征峰,2 936和2 870 cm-1左右处为碳纳米纤维的亚甲基(-CH2-)伸缩振动吸收峰,3 000~3 600 cm-1范围内为g-C3N4的N-H键伸缩振动带。而1 200~1 700 cm-1范围出现的多重强吸收峰是g-C3N4的芳香碳氮杂环结构单元、-NH和-NH2结构单元在此范围的特征峰[25]与碳纳米纤维在此范围的亚甲基(-CH2-)的C-H吸收峰、C-N键的特征吸收峰相重叠所形成。这些特征表明,制备得到的g-C3N4/C纳米纤维是由g-C3N4和含有亚甲基、C-N键的未完全碳化的碳纳米纤维构成,这是因为碳化温度仅600℃,PAN预氧化纤维含有的亚甲基和C-N键不可能完全分解形成碳化度完全的碳纳米纤维。
2.2XRD分析
g-C3N4/C纳米纤维膜的XRD图如图2所示。可以看出,在2θ为26.5°附近存在明显的特征衍射峰。PAN在碳化过程中,除碳以外的其他杂原子(H、N)以小分子(H2O、NH3)挥发的形式逐渐脱除,碳元素所占的比例逐渐增大,该处的特征峰属于石墨层的(002)衍射峰[26],这说明经600℃碳化后得到的g-C3N4/C纳米纤维膜中的碳存在石墨相。由于纤维组分中所含g-C3N4相对较少,且g-C3N4位于2θ为28.5°处的(002)衍射峰[27]处于石墨层的(002)衍射峰附近,被包含在碳的(002)衍射峰内,因此看不到单独的g-C3N4衍射峰。
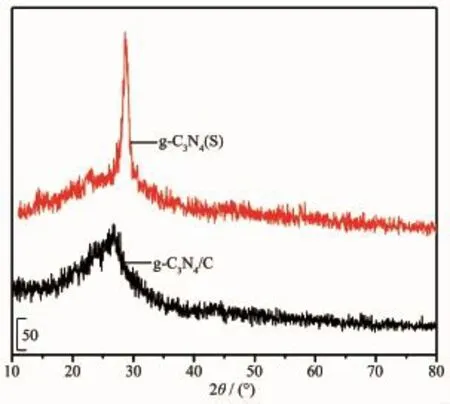
图2 g-C3N4(S)和g-C3N4/C纳米纤维膜的XRD图Fig.2 XRD patterns of g-C3N4(S)and g-C3N4/C nanofibers
2.3Raman分析
图3为g-C3N4/C纳米纤维膜的Raman谱图。从图中可以看出,Raman光谱有3个明显的谱线:1 356 cm-1附近处的D谱线,1 575 cm-1附近处的G谱线和2 806 cm-1附近处的谱线。G谱线表征的是石墨结构的完整程度,D谱线表征的是石墨结构的缺陷程度[28]。图中D线强度比G线略高,说明碳纤维有一定的石墨化程度和结构有序程度,这与XRD结果相一致,但石墨结构不完善,无定形碳原子含量较多,碳纤维主要为无定形相与石墨相混合型结构。在2 806 cm-1处的宽峰是由C-N键的振动引起[29],说明复合纤维中存在较多的C-N键,它们源于未完全碳化碳纤维和g-C3N4的C-N基团,这与FTIR分析相吻合。
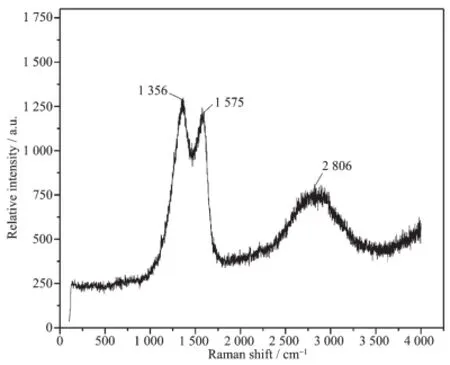
图3 g-C3N4//C纳米纤维膜的Raman光谱Fig.3 Raman spectrum of g-C3N4/C nanofibers
2.4UV-Vis DRS分析
图4为g-C3N4(S)和g-C3N4/C纳米纤维的UVVis DRS谱。可见,它们具有相似的特征,在200~400 nm波长范围内有较强的吸收,对应的是太阳光线的紫外光波长区间,吸收区域从紫外光一直延续到可见光,大于400 nm波长后,吸收强度逐渐减小,吸收带边缘约在460 nm处,波长超过460 nm后,不再具有吸收效应。这表明,g-C3N4/C纳米纤维对紫外光和可见光均具有较好的吸收特性,使其能够表现出紫外光和可见光催化降解性能。我们知道,碳纤维不具备半导体属性,对全波长光不会发生吸收,因此g-C3N4/C纳米纤维对紫外光和可见光的吸收特性源于g-C3N4,g-C3N4的存在是g-C3N4/C纳米纤维具有可见光响应的原因[30]。
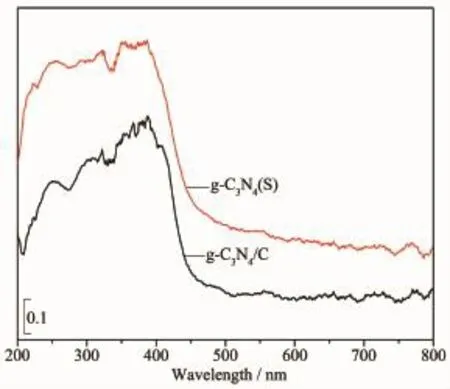
图4 g-C3N4(S)和g-C3N4/C纳米纤维膜的UV-Vis DRS图Fig.4 UV-vis DRS spectra of g-C3N4(S)and g-C3N4/C nanofibers

图5 g-C3N4/PAN(a,b)和g-C3N4/C(c,d)纳米纤维膜的SEM图Fig.5 SEM images of g-C3N4/PAN(a,b)and g-C3N4/C(c,d)nanofiber membranes
2.5表面形貌分析
图5为g-C3N4/PAN和g-C3N4/C纳米纤维膜SEM图。从2 000倍的图5(a)中可以看出,PAN与g-C3N4/C纳米片混合共纺的纤维表面较为光滑,纤维结构连续,有少量的串珠结构,说明可以达到共纺的目的。从10 000倍的图5(b)中可以看出,g-C3N4/ PAN纤维膜的直径大小主要分布在100~500 nm区间,纤维连续性好。图5(c)为经600℃碳化后所得g-C3N4/C的2 000倍SEM纤维图,可以看出,经碳化后纤维膜形状仍然保持完好,但纤维整体发生收缩,有部分纤维断裂,可能是因为g-C3N4的加入使形成的碳纤维强度降低,在碳化过程中由于纤维收缩和杂原子(H、N)产生的气体小分子(H2O、NH3)在高温挥发过程中对纤维的冲击共同导致部分纤维发生断裂。还可以发现,纳米纤维上存在少量的g-C3N4团聚颗粒,纤维直径主要分布在100~1 000 nm,表明碳化后纤维收缩,纤维直径略微变大。图5(d)为10 000倍的局部放大图,可以较明显地看出,g-C3N4纳米片粒径较小,主要在100 nm以下,大部分能够稳定良好地固定在碳纤维内部,少量分布在碳纤维表面。
2.6比表面积和孔径分布分析
从图6可以看出,g-C3N4/C纳米纤维样品的氮气吸附-脱附等温线为Ⅳ型等温线,在0.45~1.0(p/p0)范围内有明显的吸附滞后环,Ⅳ型等温线是中孔固体最普遍出现的吸附行为,结合其孔径分布图可以得出,纳米纤维之间交联形成各种孔径不一的介孔和大孔,由多点BET法计算得到纳米纤维样品的比表面积为39 m2·g-1,由孔径分布得出孔径主要分布在49.6 nm附近。可见,所纺出来的g-C3N4/C纤维内部含有少量的介孔,样品中的大孔为纤维交联所形成,它们赋予纤维较大的比表面积,这使g-C3N4/C纤维可以具有较好的光催化性能。
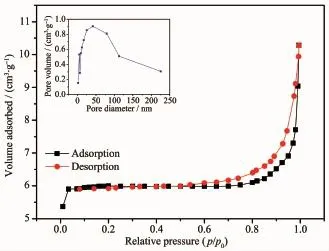
图6 g-C3N4/C纳米纤维样品的氮气吸附-脱附等温线和孔径分布图Fig.6 Nitrogen adsorption-desorption~isotherms and pore size distribution of g-C3N4/C nanofibers
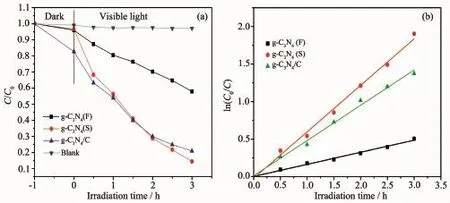
图7 (a)不同样品对RhB的可见光降解曲线;(b)光催化反应动力学拟合曲线Fig.7(a)Photodegradation curves toward RhB under visible light for various samples; (b)Compare about kinetics of RhB degradation for various samples
2.7光催化降解性能研究
图7(a)反映了g-C3N4(F)、g-C3N4(S)和g-C3N4/C纳米纤维膜光催化降解RhB效果。从空白样曲线可以看出,RhB在可见光照射下自身不发生降解。超声剥离得到的g-C3N4(S)及其电纺g-C3N4/C纳米纤维膜样品的光催化反应速率和降解率比粉末g-C3N4即g-C3N4(F)大得多。从图可见,经1 h黑暗吸附,g-C3N4(S)和g-C3N4(F)的平衡吸附量都很小(小于5%),而g-C3N4/C纳米纤维膜的吸附较明显,达到17.3%,这说明碳纤维的存在增大了样品对染料的吸附能力。3 h可见光照射后,g-C3N4(S)对RhB的降解率达到85.6%,g-C3N4/C纳米纤维膜达到79.8%,而g-C3N4(F)仅42.1%。g-C3N4(S)和g-C3N4(F)的降解率差异的原因是显然的,因为g-C3N4(S)为纳米尺寸薄片,比表面积较大,因此与待降解目标污染物RhB单位接触面积大,以致光催化降解速率远大于粉末g-C3N4(F)。让我们感到惊喜的是,g-C3N4/C纳米纤维膜按理必然有大量g-C3N4(S)被包埋在碳纤维内部和外表面上,有效比表面积必然比g-C3N4(S)小得多,光催化降解能力应该远低于g-C3N4(S),但是其与g-C3N4(S)相差竟然并不多,表现出比粉末g-C3N4(F)高得多的光催化降解能力。我们有理由推测,无定形相与石墨相共存的碳基体仍然具有比g-C3N4(S)高得多的导电能力,使暴露在纤维表面的g-C3N4(S)产生光生载流子能够快速传导,大大降低光生电子-空穴对复合的几率,以至暴露在纤维表面的并不多的g-C3N4(S)显示了很高的光催化效率。此外,g-C3N4/C纳米纤维膜周围RhB平衡浓度较高,这种浓度效应也有利于光催化。这两种因素的综合效应,使g-C3N4/C纳米纤维膜获得较高的光催化效率。从图7(b)的光催化降解RhB的动力学拟合图可以看出,g-C3N4(S)和g-C3N4/C纳米纤维膜的拟合斜率比g-C3N4(F)大,表1列出了g-C3N4(F)、g-C3N4(S)和g-C3N4/C光催化反应动力学拟合方程,通过计算可知,g-C3N4(S)样品对RhB的光催化反应速率常数是粉末g-C3N4(F)的3.85倍,而g-C3N4/C纳米纤维膜的光催化反应速率常数是粉末g-C3N4(F)的2.92倍。

表1 光催化反应速率拟合方程Table 1 Fitting equations for photocatalytic reaction rate
从反映光催化稳定性的图8可以看出,g-C3N4(S)光催化降解RhB的5次降解率依次为85.1%、66.4%、51.9%、37.5%和21.1%;g-C3N4/C纳米纤维膜对应的5次降解率依次为74.7%、68.5%、64.2%、55.3%和49.2%。通过比较可以发现,g-C3N4(S)第一次使用的催化降解效果较好,但回收后催化降解能力快速下降,到第5次时只有21.1%。这是因为g-C3N4(S)在使用过程中逐渐发生团聚,样品有效比表面积逐渐减小,同时回收时不可避免有少量损失,不能完全回收,g-C3N4(S)量有所减少,以致降解能力迅速变弱。g-C3N4/C纳米纤维膜样品首次使用的降解率为73.6%,尽管不及第一次使用g-C3N4(S)样品,但却比粉末g-C3N4(F)的38.7%有较大的提高。g-C3N4/C纳米纤维膜在使用过程中不存在g-C3N4团聚导致光催化表面减少的问题,同时碳纤维本身具有较好的韧性,在光降解过程中依然能保持比较好的膜形态,使得纤维膜样品易于回收,回收循环使用时质量损失较少,因此表现出比g-C3N4(S)样品高得多的稳定性,第5次使用仍然能保持49.2%的较高降解率,光催化稳定性较好。
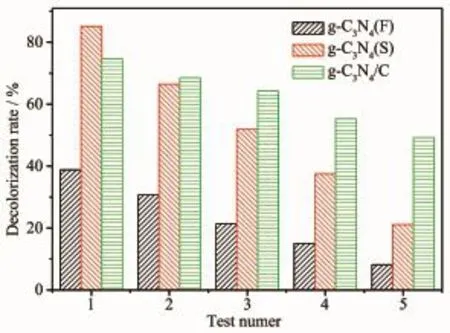
图8 g-C3N4(F)、g-C3N4(S)和g-C3N4/C纳米纤维膜5次光催化效果比较Fig.8 Compare about five photocatalytic test results of g-C3N4(F),g-C3N4(S)and g-C3N4/C nanofiber membranes
3 结论
采用PAN与g-C3N4纳米片静电混合纺丝制得的g-C3N4/C纳米复合纤维,其g-C3N4纳米片均匀地分散在复合纤维内部和表面,但存在部分团聚颗粒,复合纤维内部的碳基体存在大量的亚甲基和CN键,为含部分石墨相的无定形碳结构。无定形相与石墨相共存的碳基体能够降低纤维表面g-C3N4(S)产生的光生电子-空穴对复合的几率,使光生载流子快速传导,g-C3N4/C纳米复合纤维表现出较好的可见光催化活性,回收再用方便,循环利用稳定性良好。
[1]Wang X C,Maeda K,Thomas A,et al.Nat.Mater.,2009,8 (1):76-80
[2]CHEN Bo-Cai(陈博才),SHEN Yang(沈洋),WEI Jian-Hong (魏建红).Acta Phys.-Chim.Sin.(物理化学学报),2016,32 (6):1371-1382
[3]Cui Y J.Chin.J.Catal.,2015,36(3):372-379
[4]ZHAO Xue-Guo(赵学国),HUANG Li-Qun(黄丽群),LI Jia-Ke(李家科).Chinese J.Inorg.Chem.(无机化学学报),2015, 31(12):2343-2348
[5]Liu L,Qi Y,Lu J,et al.Appl.Catal.B:Environ.,2016,183: 133-141
[6]Liu L,QiY,Hu J,etal.Appl.Surf.Sci.,2015,351:1146-1154
[7]Bai X,Zong R,Li C,etal.Appl.Catal.B:Environ.,2014,147: 82-91
[8]Maeda K,Wang X,Nishihara Y,et al.J.Phys.Chem.C, 2009,113(12):4940-4947
[9]Hughbanks T,Tian Y.Solid State Commun.,1995,96(5):321 -325
[10]GUI Ming-Sheng(桂明生),WANG Peng-Fei(王鹏飞),YUAN Dong(袁东),et al.Chinese J.Inorg.Chem.(无机化学学报), 2013,29(10):2057-2064
[11]Niu P,Zhang L,Liu G,et al.Adv.Funct.Mater.,2012,22 (22):4763-4770.
[12]CHU Zeng-Yong(楚增勇),YUAN Bo(原博),YAN Ting-Nan (颜廷楠).J.Inorg.Mater.(无机材料学报),2014,29(8):785-794
[13]Chen L,Huang D,Ren S,etal.Nanoscale,2013,5(1):225-230
[14]Yang S,Gong Y,Zhang J,et al.Adv.Mater.,2013,25(17): 2452-2456
[15]Li D,Xia Y.Adv.Mater.,2004,16(14):1151-1170
[16]Miao J,Miyauchi M,Simmons T,et al.J.Nanosci.Nanotechnol.,2010,10(9):5507-5519
[17]LU Jian-Jian(卢建建),YING Zong-Rong(应宗荣),LIU Xin-Dong(刘信东),et al.Acta Phys-Chim.Sin.(物理化学学报), 2015,31(11):2099-2108
[18]SUN Li-Ping(孙丽萍),ZHAO Hui(赵辉),WANG Wen-Xue (王文学),et al.Chinese J.Inorg.Chem.(无机化学学报), 2014,30(4):757-762
[19]Nalbandian M,Zhang M,Sanchez J,et al.J.Mol.Catal.A: Chem.,2015,404:18-26
[20]Kim M,Kim Y,Lee K M,et al.Carbon,2016,99:607-618
[21]ZHANG Jiao-Bo(张校菠),CHEN Ming-Hai(陈名海),ZHANG Jiao-Gang(张校刚),et al.Acta Phys-Chim.Sin.(物理化学学报),2010,26(12):3169-3174
[22]Di Valentin C,Pacchioni G,Selloni A.Chem.Mater.,2005, 17(26):6656-6665
[23]Samadi M,Shivaee H A,Pourjavadi A,et al.Appl.Catal. A:Gen.,2013,466:153-160
[24]Lin Q,Li L,Liang S,et al.Appl.Catal.B:Environ.,2015, 163:135-142
[25]Lee H,Kim H,Kang S,et al.J.Ind.Eng.Chem.,2015,21: 736-740
[26]Cui Y,Tang Y,Wang X.Mater.Lett.,2015,161:197-200
[27]Liu L,Qi Y,Hu J,et al.Mater.Lett.,2015,158:278-281
[28]LI Dong-Feng(李东风),WANG Hao-Jing(王浩静),WANG Xin-Kui(王心葵).Spectrosc.Spect.Anal.(光谱学与光谱分析),2007,27(11):2249-2253
[29]Bourlinos A B,Giannelis E P,Sanakis Y,et al.Carbon, 2006,44(10):1906-1912
[30]Mousavi M,Habibi-Yangjeh A.J.Colloid Interface Sci., 2016,465:83-92
Synthesis of g-C3N4/C Nanofibers by Electrospinning and Their Photodegradation Performance under Visible Light
YANG Jia-Jia JIANG Ke-Wang LIN Xue-Mei YING Zong-Rong*ZHANG Wen-Wen
(School of Chemical Engineering,Nanjing University of Science and Technology,Nanjing 210094,China)
g-C3N4/C composite nanofibers were prepared via a combination process of electrospinning, preoxidation and carbonization by using g-C3N4nanosheets and polyacrylonitrile as raw materials.Fourier transform infrared spectrometer(FTIR),X-ray diffraction(XRD),Raman spectroscopy(Raman)and scanning electron microscopy(SEM)were employed to analyze the structure and morphology of the as-synthesized nanofibers.And UV-Vis diffuse reflectance spectroscopy(UV-Vis DRS)was used to assess their visible light response.The results show that the g-C3N4/C composite nanofibers exhibitgood photocatalytic degradation activity toward rhodamine B under visible light,which originates from better ability of their partially amorphous carbon matrix to reduce the combination of the photogenerated electron and hole pair.The nanofiber membrane was not embrittled into powers or small flakes during the photocatalytic degradation process under stirring conditions, maintaining its integrity from begin to end.After several recovery and photocatalysis experiments,the membrane still maintained high photodegradation rate.This study reveals that the resulting nanofibers have excellent recycling stability to photodegradate rhodamine B under visible light.
graphite-like carbon nitride;carbon fiber;electrospinning;composite nanofiber;visible light;photodegradation
O613.71;O643.36+1
A
1001-4861(2016)12-2088-07
10.11862/CJIC.2016.279
2016-03-28。收修改稿日期:2016-10-09。
*通信联系人。E-mail:zrying@njust.edu.cn
