肉芽肿性皮肤松弛症
2016-12-06徐丽红
陆 原,关 杨,王 鹏,李 清,柴 宝,徐丽红
肉芽肿性皮肤松弛症
陆原,关杨,王鹏,李清,柴宝,徐丽红
蕈样肉芽肿;淋巴瘤;皮肤松弛
肉芽肿性皮肤松弛症(granulomatous slack skin, GSS)是皮肤T细胞淋巴瘤中低度恶性的一型,临床罕见,现报道1例。
临床资料
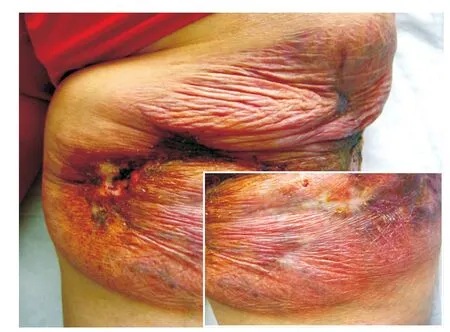
图1 肉芽肿性皮肤松弛症患者臀部皮损
患者,女,58岁。主因右臀部结节、斑块10余年,局部破溃疼痛3个月,于2014年5月22日就诊。患者10余年前无明显诱因右侧臀部反复出现大小不等的肤色或黄红色的丘疹、结节,偶有痒痛感。皮损数目逐渐增多,并有融合倾向。患者曾在多家医院就诊,曾按皮肤感染、脂膜炎、血管炎、皮肤结核等治疗(具体不详),疗效欠佳。皮损曾好转,部分甚至出现消退倾向,但继而又加重,好转与复发交替发生。5年前自觉皮损处出现皱褶,且范围逐渐扩大,触摸皮肤硬韧而无弹性,并逐渐出现皮肤下垂;曾在外院行组织病理检查,显示肉芽肿改变,口服醋酸泼尼松、雷公藤多苷等药物(剂量不详)治疗,疗效欠佳。近3个月来,皮损局部出现疼痛、破溃,有少量分泌物。患者自发病以来全身其他部位未出现肿块,无发热,体重无明显减轻。发病前无用药史及局部创伤史。既往胆囊炎病史10年、高血压病病史4年(口服降压药,血压控制良好),1年前患急性甲型肝炎,已治愈。无肺气肿、胃肠道憩室、胃下垂、直肠脱垂、腹股沟疝、泌尿系憩室、直肠脱垂、子宫脱垂等病史。无药物过敏史。父母非近亲结婚,父母、兄弟姐妹及3个子女均健康。家族中无类似疾病患者。体格检查:生命体征平稳,血压110 /70 mmHg(1 mmHg =0.133 kPa),发育正常,营养中等,双肺呼吸音低,未闻及干湿性啰音,心律齐,未闻及心脏杂音,双下肢无水肿;全身浅表淋巴结未触及增大,其他系统检查均正常。皮肤科检查:右侧臀部大片暗褐红色斑块,表面皮肤松弛下垂呈帷幔状,皮肤萎缩变薄、皮下血管清晰可见,部分皮损溃疡,无隆起性及浸润性瘢痕;皮损质地较坚实,弹性减退,无压痛(图1)。实验室及辅助检查:血常规:白细胞8. 9×109/L[正常值(4~10)×109/L],淋巴细胞百分比15. 3% (20%~40%),血红蛋白86 g/L(110~150 g/L),红细胞沉降率32 mm/1h (≤15 mm/1h),血糖6.32 m mol/L(3.9~6.1 mmol/L),抗核抗体、抗双链 DNA抗体、抗 RO抗体、抗LA抗体、SSA和 SSB抗体均阴性。类风湿因子(RF)阴性。尿、粪常规,血清电解质,肝、肾功能均正常,血脂、乳酸脱氢酶、碱性磷酸酶、磷酸肌酸激酶均正常,胸部X线、心电图均正常。腹部多谱勒超声显示肝、胰、脾、肾、胆囊均正常。皮损组织病理示表皮萎缩,真皮浅层明显水肿,真皮及皮下脂肪组织内可见灶状淋巴细胞浸润,未见明显异形性,无蕈样肉芽肿细胞及Pautrier微囊肿;浸润灶中散在组织细胞,无干酪样坏死,并见散在多核巨细胞,无吞噬现象,炎性细胞浸润区域内弹性纤维消失(图2)。免疫组化染色:CD4、CD68、CD45RO、LCA均阳性 ,CD3、CD5、CD8、CD20均阴性,ki-67阳性细胞比<10%(图3)。诊断:GSS。诊断明确后失访。
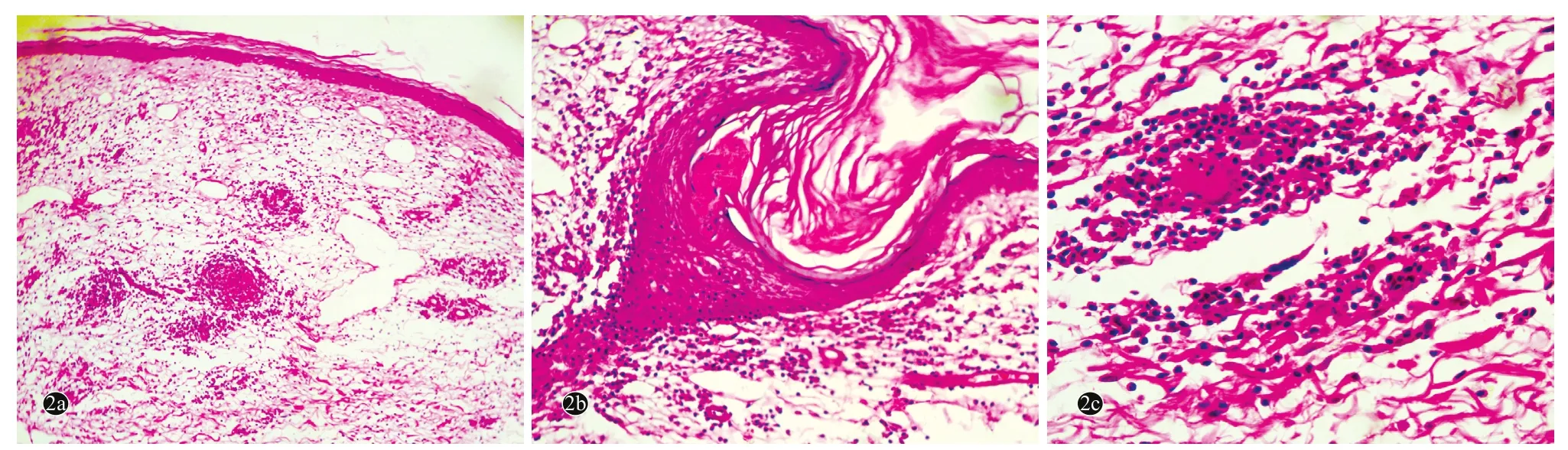
图2 肉芽肿性皮肤松弛症患者皮损组织病理(HE染色)
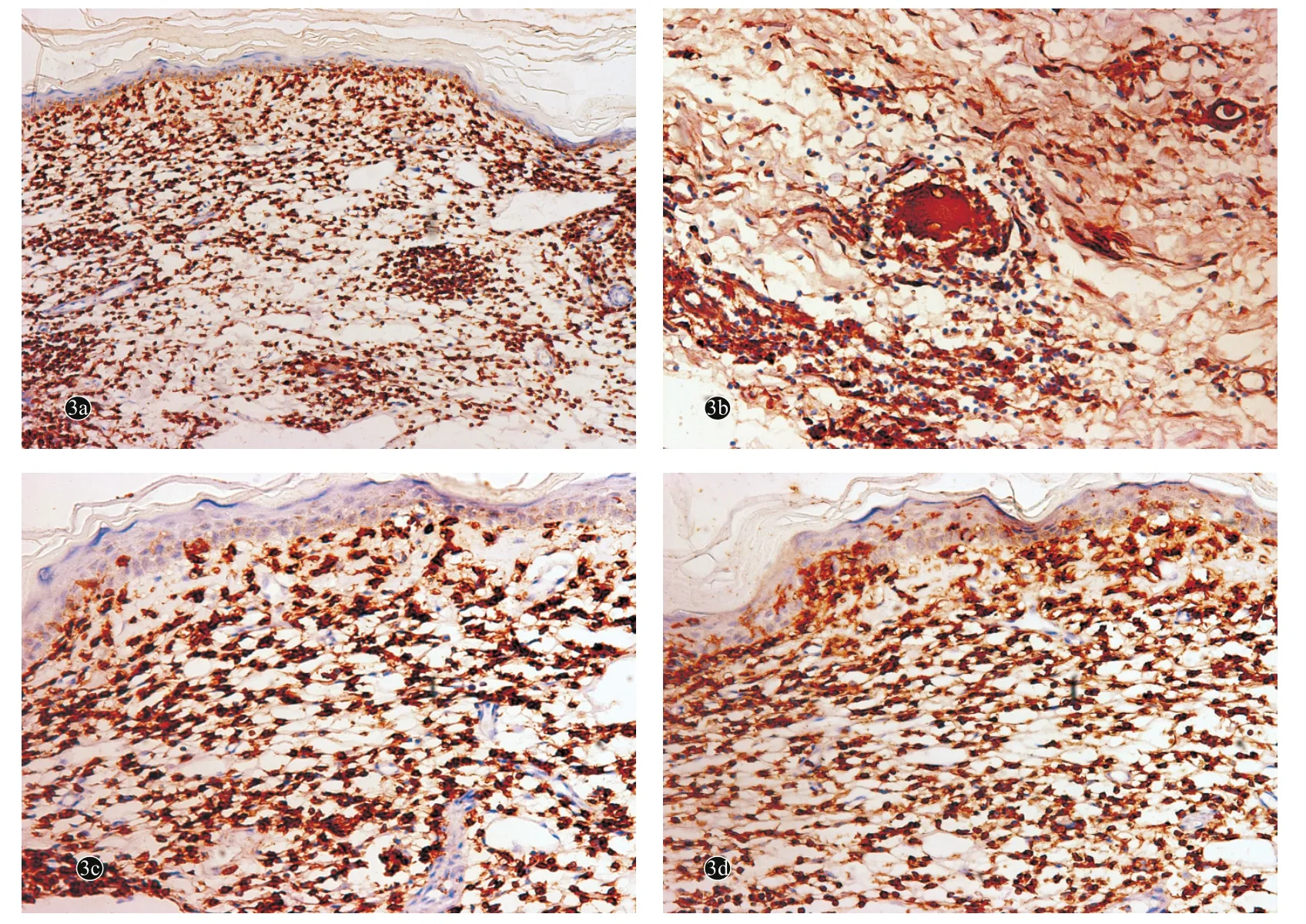
图3 肉芽肿性皮肤松弛症患者皮损免疫组化(SP法)
讨论
GSS由Convit 等[1]于1973年首先报道,但当时命名为慢性进行性萎缩性皮肤硬皮病。后Ackerman[2]总结多例患者组织病理资料,将其命名为GSS。分子生物学及细胞基因学方面的研究显示,GSS属于T细胞淋巴瘤,但GSS与其他类型T细胞淋巴瘤的不同之处在于组织病理上有弹性纤维吞噬现象。1999年,欧洲癌症治疗研究组织(EORTC)正式将GSS归类为皮肤T细胞淋巴瘤[3,4]。 近年来,关于GSS与霍奇金淋巴瘤、非霍奇金淋巴瘤及蕈样肉芽肿等恶性淋巴细胞增生性疾病的关系引起众多学者的关注,尚待定论,但普遍认为它是相对惰性的皮肤T细胞淋巴瘤[3,4]。
本病是一种极为少见的疾病,迄今能检索到有关GSS病例报道的文献仅50余例。GSS的主要临床组织病理学特征为皮肤松弛及肉芽肿形成[5]。病程慢性迁延,发病初期的临床表现缺乏特异性,发病数年后出现皮肤松弛,表现为皮肤弹性降低、起皱、悬垂及萎缩,好发部位多为皱褶处,如腋窝、腹股沟,也可以累及腹部、下肢、上肢等,也可泛发全身。多数GSS患者随着病情的进展形成垂于体表的浸润性或隆起性肿块,但也有部分患者仅表现为皮肤松弛。GSS的发病年龄跨度较大(14~69岁),但常出现在20~40岁。男性发病率略多于女性。最初报道几乎仅限于白种人,现亚洲等地的报道也日渐增多[6]。GSS很少累及皮肤以外组织,患者通常健康状况良好,但也有首发或并发蕈样肉芽肿、霍奇金淋巴瘤以及淋巴结、脾脏、支气管黏膜下受累的病例[7]。
组织病理学特点早期表现为小淋巴细胞带状浸润,没有明显核异形性。进展期病变显示致密淋巴细胞浸润全层。可见淋巴样细胞轻度异形及亲表皮性,但核异形性不及蕈样肉芽肿明显。诊断的特点是具有许多组织细胞及多核巨细胞散布在致密的淋巴细胞背景中,形成非干酪性多核巨细胞肉芽肿。部分多核巨细胞形态巨大,有10~90个细胞核,呈环形排列或聚集在细胞一侧。还可见多核巨细胞吞噬弹性纤维和淋巴样细胞现象。弹性纤维染色显示受累皮肤缺乏弹性纤维。超微结构显示淋巴细胞具有深染的脑回状细胞核,类似于蕈样肉芽肿和 Sézary 综合征[7]。肿瘤性淋巴细胞显示辅助性T细胞表型,表达CD3、CD4 、CD45RO,可丢失其他T细胞标志物,少数患者的肿瘤细胞表达CD30。多核巨细胞表达单核细胞的CD14抗原和吞噬细胞的CD68抗体,反映增生活性的Ki-67抗体阳性率为5%[3]。多数患者可见TCR 基因克隆重排,是疾病早期有用的诊断手段。有患者出现了8号染色体三体型[8]。
GSS与肉芽肿性蕈样肉芽肿(GMF)的组织病理表现及免疫表型相似。临床上GSS好发于褶皱部位,表现为皮肤松弛下垂;而GMF无特殊好发部位,常表现为丘疹或斑块而不是皮肤松弛。GMF淋巴样细胞异形性明显,常有蕈样肉芽肿细胞及Pautrier 微囊肿,多核巨细胞数目较少且胞核少(5~10个),真皮弹性纤维仅局部丢失。针对多核巨细胞,GSS与GMF鉴别在于GSS的多核巨细胞更大,所含核更多,可达20~90个,并有巨细胞吞噬弹性纤维现象及可能由此所致的弹性纤维破坏[9]。但是二者在组织病理学及临床上常有重叠[10],总体来看,GSS的愈后远比GMF要好。GSS临床上还需要和一组以皮肤起皱、松弛、萎缩及弹性纤维缺失为特征的疾病进行鉴别,如皮肤松弛症(anetoderma)以发生局限性圆形或椭圆形皮肤松弛的疝样斑为特征;先天性皮肤松弛症一般在出生时或生后不久即发生,常有家族史;获得性皮肤松弛症多继发于荨麻疹、湿疹、多发性骨髓瘤等疾病,松弛常累及颜面部,无褶皱部位受累的趋势;而真皮中层弹性纤维溶解症的皮损仅呈现波形褶皱。在组织病理上,上述疾病均无真皮内淋巴样细胞浸润及肉芽肿形成[11]。
GSS目前尚无标准治疗方案。治疗方法包括局部手术切除、放疗、补骨脂素长波紫外线(PUVA)、外用糖皮质激素制剂、系统应用糖皮质激素、化疗、α干扰素、阿维A酯、氯苯吩嗪或硫唑嘌呤等免疫抑制剂以及上述方法联合应用,但疗效均欠佳[3,12]。学者们普遍认为,更好地认识MF肿瘤细胞的生物学特征对于选择合适的治疗有重要的意义[13]。GSS的预后主要取决于其并发的恶性淋巴细胞增生性疾病的恶性程度(如:霍奇金淋巴瘤、蕈样肉芽肿、淋巴结非霍奇金淋巴瘤、白血病等),因此必须密切随访观察。
[1]Convit J, Kerdel F, Goihman M, et al. Progressive, atrophying, chronic granulomatous dermohypodermitis. Autoimmune disease? [J]. Arch Dermatol, 1973, 107(2):271-274.
[2] Ackerman AB. An algorithmic method for histologic diagnosis of inflammatory and neoplastic skin diseases by analysis of their patterns [J]. Am J Dermatopathol, 1985, 7(2):105-107.
[3] Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC) [J]. Arch Dermatol, 2008, 144(12):1609-1617.
[4] Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas [J]. Blood, 2005, 105(10):3768-3785.
[5] Balus L, Manente L, Remotti D, et al .Granulomatous slack skin: Report of case and review of the literature [J]. Am J Dermatopathol , 1996, 18(2):199-206.
[6] Pai K, Rao R, Devadiga R, et al. Granulomatous slack skin [J]. Int J Dermatol, 2000, 39(5):374-376.
[7] Clarijs M , Poot F , Laka A, et al. Granulomatous slack skin : treatment with extensive surgery and review of the literature [J]. Dermatology, 2003, 206(4):393-397.
[8] Balus L, Manente L, Remotti D, et al. Granulomatous slack skin. Report of a case and review of the literature [J]. Am J Dermatopathol, 1996, 18(2):199-206.
[9] Fischer M, Wohlrab J, Audring TH, et al. Granulomatous mycosis fungoides Report of two cases and review of the literature [J]. J Eur Acad Dermatol Venereol, 2000, 14(3):196-202.
[10] Wollina U, Graefe T, Fuller J, et al. Granulomatous slack skin or granulomatous mycosis fungoides-a case report. Complete response to percutaneous radiation and interferon alpha [J]. J Cancer Res Clin Oncol, 2002, 128(1):50-54.
[11] 赵庆利, 任建宏, 万里华, 等. 肉芽肿性皮肤松弛症 [J]. 临床皮肤科杂志, 2008, 37(10):651-653.
[12] Goldsztajn KO, Moritz Trope B, Ribeiro Lenzi ME, et al. Granulomatous slack skin. Histopathology diagnosis preceding clinical manifestations by 12 years [J]. J Dermatol Case Rep, 2012, 6(4):108-112.
[13] Lundin J, Osterborg A. Therapy for mycosis fungoides [J]. Curr Treat Options Oncol, 2004, 5(3):203-214.
(本文编辑祝贺)
Granulomatous slack skin
R739.5
B
1674-1293(2016)01-0067-03
10.11786/sypfbxzz.1674-1293.20160121
518052 深圳,广东医学院附属深圳南山医院皮肤科(陆原,关杨,王鹏,李清,柴宝,徐丽红)
陆原,主任医师,研究方向:少见、疑难皮肤性病诊治及光学在皮肤科的应用,E-mail: chfsums@163.com
(2015-07-25
2015-11-02)
