模板剂缓释法合成分子筛大单晶
2016-12-05陈飞剑林清芳高子豪沈福志梁丽丽杜红宾
陈飞剑 林清芳 高子豪 沈福志 梁丽丽 杨 贞*, 杜红宾*,
(1南京大学化学化工学院配位化学国家重点实验室,南京210023) (2蚌埠医学院化学教研室,蚌埠233030)
模板剂缓释法合成分子筛大单晶
陈飞剑1,2林清芳2高子豪1沈福志2梁丽丽2杨贞*,1杜红宾*,1
(1南京大学化学化工学院配位化学国家重点实验室,南京210023) (2蚌埠医学院化学教研室,蚌埠233030)
在分子筛的合成中,通过使用四咪唑基取代的硼类化合物(四咪唑硼钠)作为模板剂,在溶剂热条件下,成功得到了磷酸铝盐分子筛AlPO4-11的大单晶。电喷雾质谱、19F和11B NMR等研究结果表明,在溶剂热条件下四咪唑硼钠起到了缓释剂的作用,其自身经历缓慢分解,持续释放低浓度咪唑分子的过程。由其释放出来的咪唑分子起到事实上的模板作用。因其浓度较低,限制了分子筛结晶过程中晶核形成的数量,从而易于导向分子筛大单晶的生成。通过引入不同种类的四取代硼类化合物作为模板剂,这种分子筛大单晶的合成策略可潜在应用于其它分子筛材料。
模板剂;分子筛;单晶;合成
0 Introduction
Zeolitic silicates and aluminophosphates,also called molecular sieves,has been paid great attention over past several decades because of their importance in industrial applications in the fields of catalysis,ion exchange,adsorption and separation[1-3].The preparation of large,high-quality single crystals of molecular sieves is highly desirable notonly from a point of view of structural determination,but also because of their uses in studying diffusion mechanism,optical and electronic devices,etc.Several strategies have been employed to prepare large crystalsofmolecular sieves[4], including the use of F-mineralizer or chelating agents (e.g.triethanolamine)as nucleation suppressor[5],controlling the release and solubility of reactive solution species in non-aqueous media[6],through the bulkmaterial dissolution[7]or the two-silicon source techniques[8].By using thesemethods,a number of zeolitic silicate and aluminophosphate single crystals have been prepared.However,large single crystals of inorganic molecular sieves are still rare.This is particularly true for the siliceous zeolites,most of which were obtained as fine powders.Their structures were often solved by powder diffraction in combination with high resolution electron diffraction and NMR methods.Nevertheless,single-crystal diffraction data often provide the most accurate and unambiguous structure solution for a zeolite.Therefore,there is a need for new methods of preparing zeolite single crystals.
During the synthesis ofmolecular sieves,organic species,known as structure-directing agents(SDAs), are widely used as additives to direct the formation of molecular sieves with special structural topology.The stability,rigidity,shape,size,hydrophilicity,charge as well as some other properties of the SDAs all make great impact on the formation of molecular sieves. Therefore,a variety of organic molecules have been used as SDAs[9].Among these,quaternary ammonium salts are the most frequently-used SDAs for synthesizing zeolites[10-13],while organic amines ormetalamine complexes are widely used for structuredirecting the formation of aluminophosphatemolecular sieves[14-15].Recently,Xiao et al.reported one-pot synthesis of Cu-SSZ-13 zeolite by utilizing a copperamine complex as an efficient SDA[16].Neutral O-containing SDAs,e.g.18-crown-6 were succeeded in synthesis of FAU[17-18],EMT[18]and KFI[19]types of silicate zeolites.Corma et al.used P-derived organic cations to prepare zeolites ITQ-27[20]and ITQ-47[21]. Quaternary boron compounds,e.g.NaB(Im)4(Im= imidazole,Scheme 1),are a class of specialmolecules that have rigid tetrahedral structures,moderate hydrophilicity,uniform shape and suitable size,and good thermal stability,which could be candidates as SDAs for zeolite synthesis.To the best of our knowledge,there is no report on using such B-containing species as SDAs to prepare zeolites, although NaB(Im)4has been used as a ligand to form porous metal-organic frameworks with interesting topology and properties[22-24].

Scheme 1 Structure of B(Im)4-
Herein,we report a new strategy for growing zeolite single crystals by utilizing imidazolesubstituted quaternary boron compounds as SDAs. During the synthesis,the boron-imidazole compounds acted as an SDA buffer,gradually decomposed and released imidazole molecules into the synthetic gel upon thermal treatment.The latter acted as real SDAs and led to the formation of single crystals of aluminophosphate AlPO4-11(AEL).Under similar conditions,the direct use of corresponding free imidazole molecules produced only fine powders.A possible decomposition pathway of the SDAs was proposed based on electrospray ionization mass spectroscopy andmulti-nuclear NMR studies.
1 Experimental
1.1M aterials
All the chemicals except the SDAs were commercially purchased and used without further purification.The SDAswere synthesized as follows.
NaB(Im)4was synthesized according to amodified procedure developed by Barton and co-workers[25]. Typically,sodium borohydride(3.78 g,0.1 mol)and imidazole(54.5 g,0.8 mol)were mixed together in a nitrogen-flushed flask attaching to an oil bubbler,and heated by an electric jacket first to 120℃for about 0.5 h.The temperature was slowly raised to 225℃to avoid violent release of hydrogen,which wasmonitored by the oil bubbler.The reaction proceeded for about 2 h,after which the evolution of gas ceased,and plenty of precipitates appeared at the bottom of the flask. The flask was cooled to room temperature,and acetone (100mL)was added to the reactionmixture to dissolve the unreacted imidazole and byproducts,leaving the product as an off-white solid.The crude product was then recrystallized from ethanol,affording 25.0 g of pure Na[B(Im)4](82.7%yield).1H NMR(D2O):6.836 (H-5),6.983(H-4),7.270(H-2).13C NMR:121.935, 128.386,140.576.11B NMR(D2O):-0.662.ESIMS (negative ion):279m/z(B(Im)4-).
1.2Synthesis of zeolites
In a typical synthesis,0.511 g of Al(OiPr)3were dispersed into 6.207 g of ethylene glycol(EG)and stirred for1 h.Afterward,0.231 gof H3PO4(85%,w/w) was added into the above mixture with stirring for another 2 h,followed successively by 0.121 g of NaB (Im)4and 35μL of HF(40%)to give rise to a homogeneous gel with an overallmolar composition of nH3PO4∶nAl(OiPr)3∶nSDA∶nHF∶nEG=1∶1.25∶0.2∶0.4∶50.The gelwas sealed in a 15 mL Teflon-lined stainless steel autoclave and heated at 175℃for 7 d under static conditions.The final products of AlPO4-11 single crystal were ltrated and washed with distilled water and anhydrous alcohol.
1.3Characterization
Elemental analyses of C,H,and N were performed on a Elementar Vario MICRO Elemental Analyzer.The inductively coupled plasma(ICP)analysis was carried out on a Perkin-Elmer Optima 3300 DV. X-ray powder diffraction(PXRD)data were collected on a Bruker D8 Advance instrument using a Cu Kα radiation(λ=0.154 056 nm)at room temperature.Scanning electronmicroscopy(SEM)images of the products were obtained on a field emission scanning electron microanalyser(Hitachi S-4800).Solid-state NMR Spectra were recorded using magic-angle spinning(MAS)techniques at room temperature and acquired at 100.62 MHz resonance frequency using a CP-MAS sequence,with a 3.0μs1H excitation pulse, 2.0 ms contact time,5 s recycle delay and 100 kHz spectral width,using proton decoupling at 60 kHz spinal 64 during acquisition.Synthesismixtures were analyzed by liquid chromatography-electrospray ionizationmass spectrometry in positive ion mode(LC-ESIMS,ThermoQuest LCQ Duo,USA)without liquid chromatography process.Tetraethylammonium bromide (TEABr)was used as the internal standard.For the EGmixture,100μL of internal standard(mTEABr/mEG, 0.301 2 g/100 g)were added.11B NMR data were obtained on Bruker DRX 500,while19F NMR were obtained on Bruker AVANCE 400M,using coaxial NMR tube(NI5CCI-B,NORELL)with D2O for lock and shimming process.
2 Results and discussion
2.1Synthesis of AlPO4-11 crystals
Table1lists typical syntheses of aluminophosphatemolecular sieves under solvothermal conditions by using NaB(Im)4as the SDA.It isseen thatNaB(Im)4had a preferential structure-directing effect for the formation of AlPO4-11(Fig.1).AlPO4-11 was obtained in a wide range of gel compositions.Nonetheless,the crystal size of AlPO4-11 showed strong dependence on the gel composition.The amount of Al in the gel makes great impact on the crystal growth of AlPO4-11. The powders of AlPO4-11 were obtained whenis 1∶1.Larger crystals of AlPO4-11 were formed when increasing the Al content up to 1.25 under otherwise identical conditions.The presence of F-is essential for the AlPO4-11 formation.When HF wasabsent or replaced by HCl,an amorphous phase was formed.Large single crystals of AlPO4-11 were formed with aratio of 0.2~0.4(Fig.2and Fig.3). Further increase of HF led to the formation of AlPO4-11 powders.The pH of the gel also played an important role in the crystallization of AlPO4-11. When NH4Fwas used instead of HF,amorphous phase was obtained and attempts to obtain AlPO4-11 failed. Moreover,the mixing sequences of the chemicals during the preparation of the synthesis gel had influence on the crystal growth.The sequence of EG→Al(OiPr)3→H3PO4→NaB(Im)4→HFwith a ratio of 1∶1.25∶0.2∶0.4∶50 led to the formation of large AlPO4-11 crystals,while changing the feed sequence to EG→Al(OiPr)3→SDA→H3PO4→HFor EG→NaB(Im)4→Al(OiPr)3→H3PO4→HF with the same ratios resulted in the formation of fine powders of AlPO4-11. Water content has great influence on the crystallization of AlPO4-11 under hydrothermal conditions[26], however,it was not taken into consideration here under solvothermal conditions.
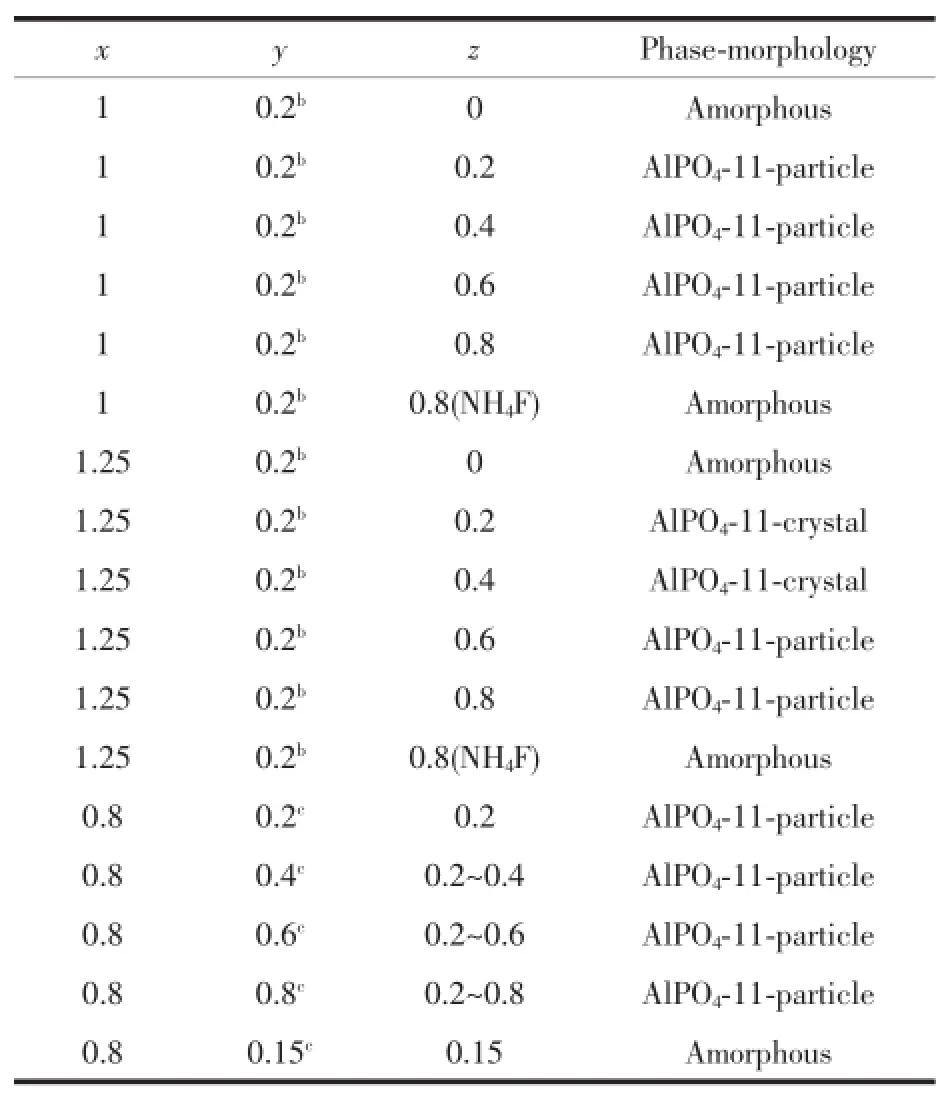
Table1 Results for the syn thesis of alum inophosphatesa
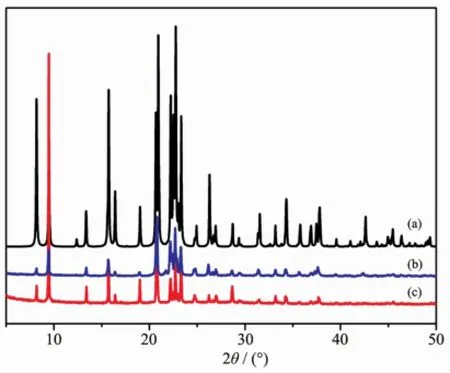
Fig.1 XRD patterns of AlPO4-11:(a)Simulated,and made with(b)Im,and(c)NaB(Im)4as SDAs

Fig.2 SEM images of differentmorphology of APO-11 made with(a1 and a2)NaB(Im)4and(b1 and b2) Im as SDAs

Fig.3 XRD patterns of AlPO4-11 with differentamountof HF(nH3PO4∶nAl(OiPr)3∶nNaB(Im)4∶nHF∶nEG=1∶1.25∶0.2∶z∶50, 175℃,7 d):z=(a)0.2(b)0.4(c)0.6(d)0.8
AlPO4-11 is a one-dimensional-pore aluminophosphate zeolite with the biggest pore of 10-memberedring running along the[001]direction[27].The channelis shaped 0.4 nm×0.65 nm in dimensions,which is too small to allow a large-sizemolecule of SDA NaB(Im)4to fill inside.Furthermore,the elemental analyses for C,H,and N shows a nC/nNratio of 1.5 for the assynthesised AlPO4-11,while there was no B element by ICP analyses.It is apparent that the intended SDA NaB(Im)4was decomposed during the solvothermal treatment and the resulting imidazole acted as SDA for the formation of AlPO4-11,which was proved by13CNMR studies of the as-synthesized samples(Fig.4).

Fig.4 Solid-state13C MASNMR spectrum of(a)as-made AlPO4-11 and(b)13C NMR spectrum of HIm+in D2O solution
Further experiments were carried out with imidazole directly used as SDA for solvothermal synthesis of AlPO4-11 under similar conditions(Table1).Not surprisingly,AlPO4-11 was obtained in each trial.It is noted,however,that the products obtained from a wide range of gel compositions are usually powders aggregates by fine crystals.Since all large crystals or powders shows nearly the same apparent morphology,only their typical SEM images are depicted in Fig.2.In addition,it was observed that high Al contentled to the formation of AlPO4-11 powders with decreased crystallinity,while better crystallized AlPO4-11 samples were obtained at lower Al content(1∶1)(Fig.2b).These are different from those obtained with NaB(Im)4as SDA,whereas the crystal size of AlPO4-11 showed strong dependence on the gel composition and large single crystalswere obtained from optimized synthetic conditions,e.g.moderate HF concentration,high Al content,and appropriate chemical-mixing sequence.
The above observations suggest that the decomposition process of NaB(Im)4playsa crucial role in the synthesis of AlPO4-11 single crystals.We hypothesize that during the solvothermal synthesis of AlPO4-11, NaB(Im)4acts as an SDA buffer,continuously supplying SDA imidazole molecules for the nucleation of AlPO4-11 and growth of large crystals.Initially,the concentration of imidazolemolecules is low,leading to the formation of only a small number of AlPO4-11 crystal nuclei.As the crystallization continues,the gradual decomposition of NaB(Im)4releases more imidazole molecules,which provides necessary SDA molecules for the growth of large AlPO4-11 crystals.
2.2Decom position of NaB(Im)4
To validate the above hypothesis,several experiments were carried out to find out how the NaB (Im)4decomposes.These included11B,19F NMR and ESIMS studies of the mixtures of NaB(Im)4in EG, NaB(Im)4in EG in the presence of different additives like HF,HCl,NH4F and NaF,respectively,and an AlPO4-11 synthetic mixture.Before taking the spectral measurements,the mixtures were solvothermally treated at 175℃for a duration of 1 h to 7 d,similar to that of AlPO4-11 synthesis.Table2lists the details of the experiments,and the results are shown in Fig. 5~8,respectively.

Fig.5 11B NMR spectra of themixture A1,A3,A6,B and B(Im)4-

Table2 Different solutions of NaB(Im)4hydrothermally treated at 175℃for 3 h*
The solution of NaB(Im)4in EG(A1):When the solution of NaB(Im)4in EG was solvothermally treated at175℃for 3 h,11BNMR and ESIMSspectra showed thatNaB(Im)4completely decomposed.The11B chemical shift at-0.41 owing to Bim4-disappeared,replaced by a new,broad peak centered at ca.8.83(Fig.5). Consistently,the negative ion peak of B(Im)4-in ESI MS spectra disappeared,suggesting that B(Im)4-was totally decomposed.In the meanwhile,the ESI MS positive ion peak of m/z 69,belonging to protonated imidazolium cation,was very weak compared to that of m/z 130 of the added internal standard TEA+(Fig.7, A1).These results suggest that there was only little amount of free imidazolium released into the solution during the decomposition of B(Im)4-.The decomposition of B(Im)4-likely resulted in the formation of some unknown polymeric boron-containing species(B), which could contribute to several observed positive ion peaks with much higher m/z in the ESI MS spectra,and the broad peak centered at 8.83 in the11B NMR spectra.

Fig.6 11B NMR spectra of themixture A2,A3,A4 and A5
The solution of NaB(Im)4with HF(A2~A5):When HF was added to the solution of NaB(Im)4in EG,the decomposition behavior of B(Im)4-under solvothermal condition was quite different to that without HF(A1). As shown in Fig.5,there were five peaks in the11B NMR spectra of the solution(A3)with amolar ratio of 1∶2,which could be assigned to BF4-(-1.79),-BF3-(-0.30),=BF2-(0.99),≡BF-(4.98)and unknown polymeric boron species B(8.5)similar to thatof A1.
The intensities of these peaks,i.e.the quantities of the corresponding species,were correlated to the added HF concentration.As shown in Fig.6,when the HF concentration was very low(0.2 as in A2),there was only a small peak around 4.98 assigned to≡BF-and a broad strong peak around 8.50 owing to polymeric boron species B.When increasing the amount of HF from 0.2 to 0.8,the11B NMR peak of species B was highly reduced,while the peaks due to the multiple F-substituted B species BF4-,-BF3-and =BF2-significantly increased.This is within the expectation sincemore F-would result in the formation of moremultiple F-substituted B species.The19F chemical shifts of these F-containing boron species were found at ca.-152.00(BF4-),-151.52(-BF3-),-150.76(=BF2-) and-150.17(≡BF-),respectively(Fig.8).Additionally, there was a small peak around-145.12 and a broad peak at ca.-133.08 in the19F NMR spectra.The former could be due to the F-substituted EG or Im, while the latter could be attributed to the mobile F-associated with polymeric species in the system.The above results suggest that the presence of F-resultedin the dissociation of B(im)4-via F-substitutions of ligand Im.This would gradually release free Im molecules into the solution.Indeed,the appearance of strong m/z peak at 69 in the ESI MS spectra confirmed the presence of large amount of imidazolium cations in the solution(Fig.7).
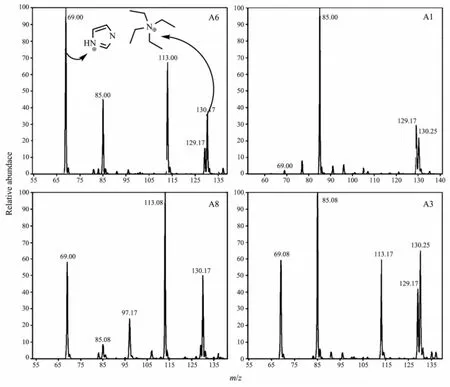
Fig.7 ESIdetecting the composition of NaB(Im)4
The solution of NaB(Im)4with only H+(A6)or F-(A7 and A8):To clarify the roles of F-and H+during the solvothermal decomposition of B(Im)4-,additional experiments were carried out.The system of A6 is the solution of NaB(Im)4-EG-HCl,in which HClwas added instead of HF.The11B NMR spectra of A6 showed that,similar to those of A1(NaB(Im)4-EG),the11BNMR peak of B(Im)4-disappeared because of its complete decomposition,and a broad peak attributable to polymeric boron species(B)appeared at ca.9.21(Fig. 5,A6).The ESIMS spectra revealed the appear-ance of a very strong m/z peak at 69 owing to the imidazolium cation,suggesting that there were large amount of free imidazolium cations released during the decomposition of B(Im)4-.The decomposition behavior of NaB(Im)4is similar to that of A3 with the presence of HF,but in contrast with those of A1 withoutH+,where therewasalmostno free imidazolium released into the solution during the decomposition of NaB(Im)4.It is noted,however,that the relative concentration of free imidazolium vs internal standard TEA+in A6 wasmuch larger than thatof A3,suggesting that strong acid HCl is more prone to decomposeB(Im)4-than HF.

Fig.8 19FNMR spectrum of themixture A3
The systems of A7 and A8 are the solutions of NaB(Im)4in EG with NaF and NH4F,respectively,in which F-ions were present but not H+.The decomposition behaviors of B(Im)4-for both systems under solvothermal condition are very similar to that of A3 with the presence of HF.Taking A8 as example(Fig. 5~7),the11B and19FNMR spectra showed the existence of BF4-,-BF3-,=BF2-,≡BF-,and unknown polymeric boron species B.Meanwhile,the ESI-MS spectra revealed the presence of large amount of free imidazolium cations released during the decomposition of B(Im)4-.In comparison with A3 or A6,the free imidazolium cations in the solution of A8 were less in quantity.These results suggest that F-alone could reactwith Bim4-and release free imidazolemolecules.
Proposed solvothermal decomposition mechanism for NaBIm4:From the above experiments,it is shown that NaB(Im)4is not stable and decomposes under solvothermal treatment at 175℃.It does not produce free Im in pure EG solution upon decomposition, while the presence of H+and/or F-promotes the release of free Im.Possible decomposition pathways were proposed,which is depicted in Fig.9.In the presence of F-as in the systems of A2~A5,A7 and A8,because F-is more nucleophilic and less steric than imidazole and H2O,it can replace the imidazole in B(Im)4-under solvothermal condition to form B(Im)3F with the releases of one imidazole at the same time. The resulting B(Im)3F is more easily attacked by additional F-,thus gradually releasingmore imidazole and forming multiple F-substituted boron imidazole complexes,and eventually BF4-.(The F-substituted boron complexes could be easily hydrolyzed;and thus these species were not detected by ESIMS studies.) On the other hand,the imidazole in BIm4-is partially nucleophilic,which could be protonated to form a neutral species B(Im)4H in the presence of H+.The latter is not thermally stable and more readily subject to nucleophilic attacks.Therefore,in the presence of H+butwithoutF-(like the caseofA6),thenucleophilic attackson B(Im)4-could occurwith weaker nucleophilic H2O to form intermediate boron acid complexes and release free imidazoles.However,in the absence of both H+and F-,such nucleophilic attacks on B(Im)4-could not happen,instead other decompositionpathways involving polymerization took place,with little amountof imidazoles released.

Fig.9 Proposed mechanism of the decomposition of NaB(Im)4in EG
2.3Structure-direction in the synthesis of AlPO4-11 crystals
The thermal behaviors of NaB(Im)4during the solvothermal synthesis of AlPO4-11 are similar to the above model systems.The gradual release of Im from B(Im)4-in the synthetic mixture of AlPO4-11 was confirmed by ESI MS studies.Since the sample appeared obvious peaks of AlPO4-11 by XRD analysis after thermal treatment for about 4 d,we thoroughly studied the decomposition behaviour of NaB(Im)4by thermal treatmentwith a duration time from 1 d to 7 d.
As shown in Fig.10,there was considerable amount of free imidazolium cations in the mixture of nH3PO4∶nAl(OiPr)3∶nSDA∶nHF∶nEG=1∶1.25∶0.2∶0.4∶50 after 1 d of solvothermal treatment.The concentration of the free imidazolium cations gradually increased with prolonged solvothermal treatment.In comparison,the concentration of the free imidazolium cations in the synthetic mixture of=1∶1.25∶ 0.8∶0.4∶50,where Im directly used as SDA wasmuch higher under the same conditions.This validates the hypothesis that we proposed earlier.That is,NaB(Im)4acts as an SDA buffer during the solvothermal synthesis of AlPO4-11,initially releasing low concentration of free SDA imidazole molecules and continuously supplying them for the formation of AlPO4-11 and growth of large crystals.Therefore,large crystals of AlPO4-11 were obtained with NaB(Im)4as SDA,while fine powders of AlPO4-11 were the main productswhen imidazole was directly used as SDA.

Fig.10 Relative abundance(m/z=69,m/z=130)of protonated imidazole after treated for different time;SDA used:(a)Im(b)NaB(Im)4
The afore-mentioned solvothermal decomposition behaviour of NaB(im)4could also account for the effects of the HF concentration in the gel and the mixing sequences on the crystal growth of AlPO4-11. Since F-ionswere incorporated into the framework of AlPO4-11,and locating in the 4-membered ring channels,these F-ions can be deemed as a co-SDA for the formation of AlPO4-11,similar to those found in the synthesis of some porous germanosilicates[28], where the F ions are known to direct the formation of the double-4-membered rings.The appropriate amount of HF concentration)is beneficial to the growth of large crystals of AlPO4-11 because both H+and F-would result in the decomposition of B(Im)4-and the release of free imidazole. Because F-reacted with B(Im)4-to form various boron complexes,the F-concentration is reduced.The lowconcentration free imidazole and F-thus acted as co-SDAs to induce the formation of large AlPO4-11 crystals.Further increase in HF concentration(nHF/ nH3PO4=0.6~0.8,Table1),however,would result in the release of large amount of imidazole and excess F-in the synthesismixture,thus leading to the formation of small crystals.In comparison to the solvothermal synthesis of AlPO4-11 with imidazole as SDA,the increase of HF concentration in the gel increased the concentration of co-SDA F-.Therefore,fine powders of AlPO4-11 with better crystallinity were obtained with higher amountof HF in the gel(nHF/nH3PO4=0.8).
The afore-mentioned studies also showed that the presence of strong acid would result in quick decomposition of B(Im)4-and release of large amount of free imidazole.Therefore,it is important to control the acidity in the synthetic mixture for the formation of AlPO4-11 crystals when NaB(Im)4is used as SDA. During the preparation of the synthetic mixture,when NaB(Im)4was added before H3PO4,as in the cases with the feed sequence of EG→Al(OiPr)3→SDA→H3PO4→HF or EG→SDA→Al(OiPr)3→H3PO4→HF, the subsequently-added strong acid H3PO4wouldimmediately protonate the imidazole in B(Im)4-.As a result,B(Im)4-quickly decomposed at the initial stage and released large amount of free imidazole upon solvothermal treatment.This led to the formation of fine powders of AlPO4-11,similar to those syntheses with imidazole as SDA.However,when H3PO4was added before addition of NaB(Im)4,as in the sequence of EG→Al(OiPr)3→H3PO4→SDA→HF,it could react with Al(OiPr)3,significantly reducing the concentration of H+.The decomposition rate of B(Im)4-was reduced and the resulting low-concentration free imidazole thus induced the formation of large AlPO4-11 crystals.
Further observations of the relationship between the Al(OiPr)3content in the gel and the resulting AlPO4-11 crystals corroborate the above reasonings: Higherin the gelmixture would resultin lower H+concentration and less free imidazole decomposed from B(Im)4-,thus forming large AlPO4-11 crystals.Indeed,large single crystals of AlPO4-11 were formedwhen Al(OiPr)3/H3PO4ratio is1.25∶1,whileAlPO4-11 powderswere obtained at nAl(OiPr)3/nH3PO4=1∶1.
3 Conclusions
We used imidazole-substituted quaternary boron compounds NaB(Im)4as SDAs in the zeolite synthesis, and successfully obtained large single crystals of AlPO4-11.These quaternary boron compounds acted as an SDA buffer,undergoing the decomposition process to slowly release a relatively low concentration of imidazole molecules under solvothermal conditions. The latter served as SDAs to form a small number of zeolite nuclei and thus lead to the growth into large single crystals.This approach could be potential a general,applicable strategy for synthesizing large crystal of other zeolites.We have extended thismethod to the formation of large single crystal of germanosilicate ITQ-12 by using such a SDA buffer and successfully obtained a more accurate and unambiguous structure solution that was different from any other results of the reference.The results will be published elsewhere.
References:
[1]Davis M E.Nature,2002,417(6891):813-821
[2]Du H,Fairbridge C,Yang H,et al.Appl.Catal.A,2005, 294:1-21
[3]Corma A,Iborra S,Velty A.Chem.Rev.,2007,107(6):2411-2502
[4]Lethbridge ZA D,W illiams JJ,Walton R I,etal.Microporous Mesoporous Mater.,2005,79(1/2/3):339-352
[5]Charnell JF.J.Cryst.Growth,1971,8(3):291-294
[6]Kuperman A,Nadimi S,Oliver S,et al.Nature,1993,365 (6443):239-242
[7]Shim izu S,Hamada H.Angew.Chem.Int.Ed.,1999,38(18): 2725-2727
[8]Sun Y,Song T,Qiu S,etal.Zeolites,1995,15(8):745-753
[9]Moliner M,Rey F,Corma A.Angew.Chem.Int.Ed.,2013, 52(52):13880-13889
[10]Burton A W,Zones S I,Elomari S.Curr.Opin.Colloid Interface Sci.,2005,10(5/6):211-219
[11]Lobo R,Zones S,DavisM.J.Inclusion Phenom.Macrocyclic Chem.,1995,21(1/2/3/4):47-78
[12]Jackowski A,Zones S I,Hwang S J,et al.J.Am.Chem. Soc.,2009,131(3):1092-1100
[13]Archer R H,Zones S I,DavisM E.MicroporousMesoporous Mater.,2010,130(1/2/3):255-265
[14]Yu J,Xu R.Chem.Soc.Rev.,2006,35(7):593-604
[15]Yu J,Xu R.Acc.Chem.Res.,2003,36(7):481-490
[16]Ren L,Zhu L,Yang C,etal.Chem.Commun.,2011,47(35): 9789-9791
[17]Delprato F,Delmotte L,Guth J L,et al.Zeolites,1990,10 (6):546-552
[18]Dougnier F,Patarin J,Guth JL,et al.Zeolites,1992,12(2): 160-166
[19]Chatelain T,Patarin J,FarréR,et al.Zeolites,1996,17(4): 328-333
[20]Dorset D L,Kennedy G J,Strohmaier K G,et al.J.Am. Chem.Soc.,2006,128(27):8862-8867
[21]Simancas R,Dari D,Velamazán N,et al.Science,2010,330 (6008):1219-1222
[22]Hamilton B H,Kelly K A,Wagler T A,et al.Inorg.Chem., 2002,41(20):4984-4986
[23]Bennett T D,Tan JC,Moggach SA,et al.Chem.Eur.J., 2010,16(35):10684-10690
[24]Chen S,Zhang J,Wu T,et al.Dalton Trans.,2010,39(3): 697-699
[25]Hamilton B H,Kelly K A,MalasiW,et al.Inorg.Chem., 2003,42(9):3067-3073
[26]Zhang B,Xu J,Fan F,etal.Microporous Mesoporous Mater., 2012,147:212-221
[27]Bennett JM,Richardson Jr JW,Pluth J J,et al.Zeolites, 1987,7(2):160-162
[28]Sastre G,Vidal-Moya JA,Blasco T.Angew.Chem.Int.Ed., 2002,41(24):4722-4726
Synthesis of Large Zeolite Crystals Through Slow Release of Structure-Directing Agents
CHEN Fei-Jian1,2LINQing-Fang2GAO Zi-Hao1SHEN Fu-Zhi2LIANG Li-Li2YANG Zhen*,1DU Hong-Bin*,1
(1State Key Laboratory of Coordination Chemistry,School of Chemistry and Chemical Engineering, Nanjing University,Nanjing 210023,China)
(2Department of Chem istry,Bengbu Medical College,Bengbu,Anhui 233030,China)
By using imidazole-substituted quaternary boron compound NaB(Im)4(Im=imidazole)as a structure directing agent(SDA)in the zeolite synthesis,we successfully obtained single crystals of aluminophosphate AlPO4-11 under solvothermal conditions.Electrospray ionization mass spectrometry,19F and11B NMR studies showed that these quaternary boron compounds acted as SDA buffers,undergoing the decomposition process to slowly release a low concentration of imidazole molecules under solvothermal conditions.The latter served as SDAs to form a limited number of zeolite nuclei,thus leading to the growth into large single crystals.Under similar conditions,the direct use of the corresponding free imidazole molecules in the synthesis produced only fine powders.This approach could be potentially applied to synthesize large crystals of other zeolites,given the availability of various quaternary boron compounds.
structure-directing agents;zeolite;single crystal;synthesis
O613.8+1
A
1001-4861(2016)07-1283-10
10.11862/CJIC.2016.168
2016-03-15。收修改稿日期:2016-05-23。
国家自然科学基金(No.21071075)、配位化学国家重点实验室开放课题计划(No.SKLCC201507)、安徽高校自然科学研究项目(No.KJ2016A462,KJ2016A463)和蚌埠医学院自然科学基金项目(No.BYKC1401ZD,BYKY1433)资助。
*通信联系人。E-mail:yangzhen@nju.edu.cn,hbdu@nju.edu.cn;会员登记号:S06N8543M1006(杜红宾)。
