先天性巨结肠患儿平面细胞极性通路核心基因的突变筛查
2016-11-25蔚开慧
苏 琳,姜 茜,蔚开慧,李 颀,张 震,肖 萍,姜 宏
先天性巨结肠患儿平面细胞极性通路核心基因的突变筛查
苏 琳1,姜 茜2,蔚开慧3,李 颀4,张 震4,肖 萍5,姜 宏1
目的 探讨平面细胞极性(PCP)信号通路核心基因突变在人类先天性巨结肠(HSCR)疾病发生中的作用。方法 收集83例HSCR患儿外周血,提取基因组DNA,对PCP通路的核心基因(CELSR3、FZD3、VANGL1、VANGL2、PRICKLE1、PRICKLE2、DVL1、DVL2)进行目标区域捕获及二代测序,针对检出的可疑致病性突变进行PCR扩增及Sanger测序验证,通过GeneTool 软件及生物信息学网站进行数据分析。结果 83例患儿中,9例患儿存在PCP通路核心基因突变,阳性率为10.8%,突变的核心基因分别为:CELSR3基因c.7724A>G(H2575R)、c.6613G>A(A2205T)、c.1961C>T (T654M)、c.2230G>A(V744M) 和 c.8615C>G(A2872G);PRICKLE1基因c.113C>T(P38L)和c.797C>T(T266I) ;DVL2 基因 c.319C>T(R107W )和c.1276G>T(V426L),数据分析提示上述突变为有害突变。结论 PCP通路核心基因突变导致的编码蛋白功能异常可能参与了HSCR疾病的发生。
先天性巨结肠;平面细胞极性通路;基因突变;CELSR3
先天性巨结肠(Hirschsprung disease,HSCR)是一种多基因、多因素参与的神经嵴细胞源性疾病[1],属于典型的肠神经系统(rnteric nervous system,ENS)先天发育异常性疾病,是导致功能性肠梗阻最常见的小儿消化道畸形。其发病率存在种族差异,欧洲人群约为15/100 000,亚洲人群最高,我国属于高发病率国家[2]。目前研究[3-4]显示并确定的14个基因的累积突变仅能解释不足20%患者的发病原因,提示其他致病基因或危险因子可能参与了HSCR的发病。PCP通路在进化上非常保守,从果蝇到哺乳动物都包括一组共同的核心基因,在脊椎动物中主要包括Fzd3(Frizzled3)、Celsr3(fmi)、Vangl、Dvl和Prickle等。其在原肠胚形成、轴突导向以及树突形成等过程中均发挥重要作用[5]。有文献[6]报道,Celsr3和Fzd3基因中任何一个缺失均可导致ENS轴突导向紊乱和胃肠道蠕动异常,症状与人类HSCR高度相似。但目前尚无直接证据证明PCP通路核心基因异常与人类HSCR的发生具有相关性。该研究旨在探讨HSCR 患者PCP通路核心基因的突变频率、类型以及相应临床表型,为进一步阐明PCP信号通路在人类ENS发育及HSCR发病中的作用,并提供相应证据。
1 材料与方法
1.1 病例资料 选择2013年4月~2015年5月在首都儿科研究所附属儿童医院普通外科诊治的83例HSCR患儿作为研究对象,其中男51例,女32例;年龄10 d~10.8岁(1.17±1.77)岁,除1例有HSCR家族史外,其余均为散发病例,其中全结肠型HSCR 32例,长段型HSCR 19例,短段型HSCR 32例。
1.2 方法
1.2.1 PCP通路核心基因捕获及二代测序 利用目标区域捕获、测序法对8个PCP通路核心基因(CELSR3、FZD3、VANGL1、VANGL2、PRICKLE1、PRICKLE2、DVL1和DVL2)的外显子及外显子-内含子交接区进行检测。
1.2.2 DNA提取及全基因组文库制备 应用德国Qiagen公司试剂盒(the QIAamp DNA Blood Midi Kit)提取患儿静脉血基因组DNA,随机打断(Biorupter),纯化长度150~250 bp之间的片段,T4 DNA Polymerase、T4 phosphorylated polynucleotide kinase和EseheriehiacoliDNA聚合酶Klenow片段对纯化后的DNA片段进行末端修复,按照美国Illumina公司二代测序仪的操作说明在片段两端加上A碱基及接头(adaptor),磁珠纯化。
1.2.3 杂交 对纯化后的模板进行PCR扩增,将PCR产物与自主设计的特异性探针(GenCap Custom Enrichment Kit,北京迈基诺基因科技有限责任公司)杂交22 h,目标片段因与特异性探针结合而被捕获,洗脱未杂交的片段,保留在探针上的目标片段再进行次行捕获PCR以显著增加目标片段的数量。
1.2.4 生物信息学分析 本实验中使用的芯片捕获区包含8个PCP通路相关基因的外显子及其侧翼序列100 bp。Illumina Pipeline(version 1.8.2)获取高通量测序原始数据,去除低质量的数据后利用BWA(Burrows wheeler aligner)将“干净的”读序与人类基因组参考序列比对(UCSC, hg19),再分别通过SOAPsnp和GATK软件收集SNP和InDel。本实验中样品基因的平均测序深度约为266.8×,捕获区覆盖度达95%以上。参考dbSNP数据库、Hapmap、千人基因组数据库及ESP(NHLBI Exome Sequencing Project)正常对照人群数据库,找出致病性点突变,将频率小于0.05的变异视为可疑。
1.2.5 PCP通路核心基因罕见突变的验证 应用在线PRIMER 3软件对序列比对发现的突变位点逐一设计引物(表1)。PCR扩增,反应体系(50 μl)为:10 ng/μl DNA模板15 μl、20 μmol/L上、下游引物各2 μl、10×PCR buffer 5 μl、2.5 mmol/L dNTPmix 5 μl、Takara Taq酶0.5 μl、去离子水20.5 μl。反应条件为94 ℃预变性1 min,35个循环(94 ℃变性30 s,55 ℃退火30 s,68 ℃延伸30 s),72 ℃延伸1 min。PCR扩增产物纯化,双向测序(北京诺赛基因组研究中心有限公司),突变位点分析。
2 结果
2.1 PCP通路核心基因突变分析 83例HSCR患儿中,9个患儿中共检出3个基因的9个错义突变,全部为杂合突变(图1),阳性率为10.8%。6例患儿发现CELSR3基因5个错义突变位点,分别为:c.7724A> (H2575R)、c.6613G>A(A2205T)、c.1961C>T(T654M)、c.2230G>A(V744M)、c.8615C>G(A2872G),其中2例患儿(HSCR0031和HSCR0048)同时出现c.1961C>T(T654M)错义突变,且突变位点位于CELSR3基因编码蛋白的一个Cadherin功能域(图2),其临床类型分别为长段型和全结肠型巨结肠(巨结肠合并胆总管囊肿),病情均较严重。 2例患儿PRICKLE1基因出现c.113C>T(P38L)和c.797C>T(T266I) 2个错义突变,另2例患儿DVL2基因出现c.319C>T(R107W)和c.1276G>T(V426L)2个错义突变。9例患儿中1例(HSCR0051)PCP通路两个核心基因CELSR3和PRICKLE1同时发生错义突变,突变位点分别为 c.2230G>A(V744M)和c.797C>T(T266I),其携带的c. 2230G > A( V744M) 突变位点也位于CELSR3 基因编码蛋白的Cadherin 功能域,该患儿临床分类为长段型巨结肠,病情严重。Sanger 测序法的验证结果与2 代测序完全吻合。全部错义突变位点的信息见表2。
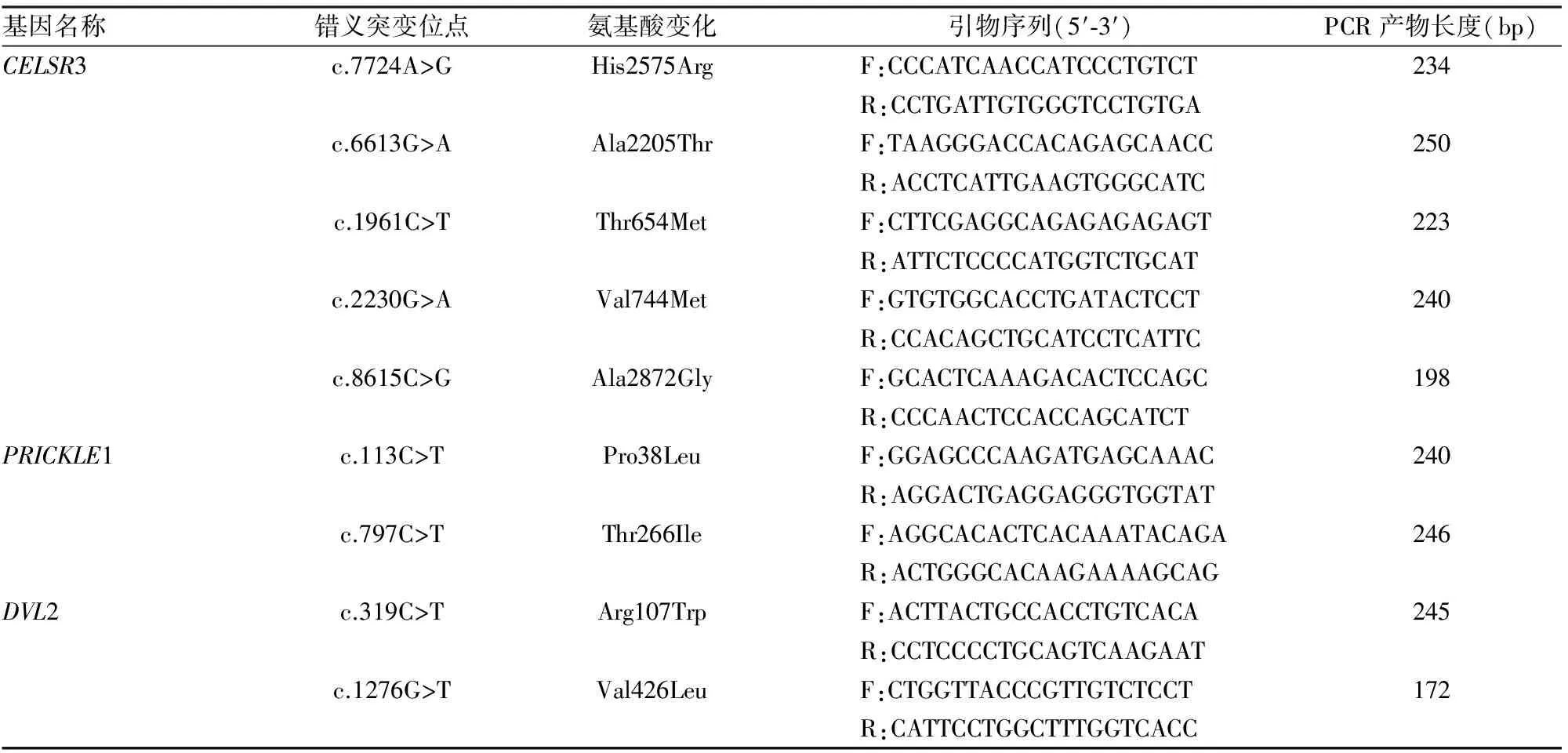
表1 PCP通路核心基因罕见突变位点验证引物信息
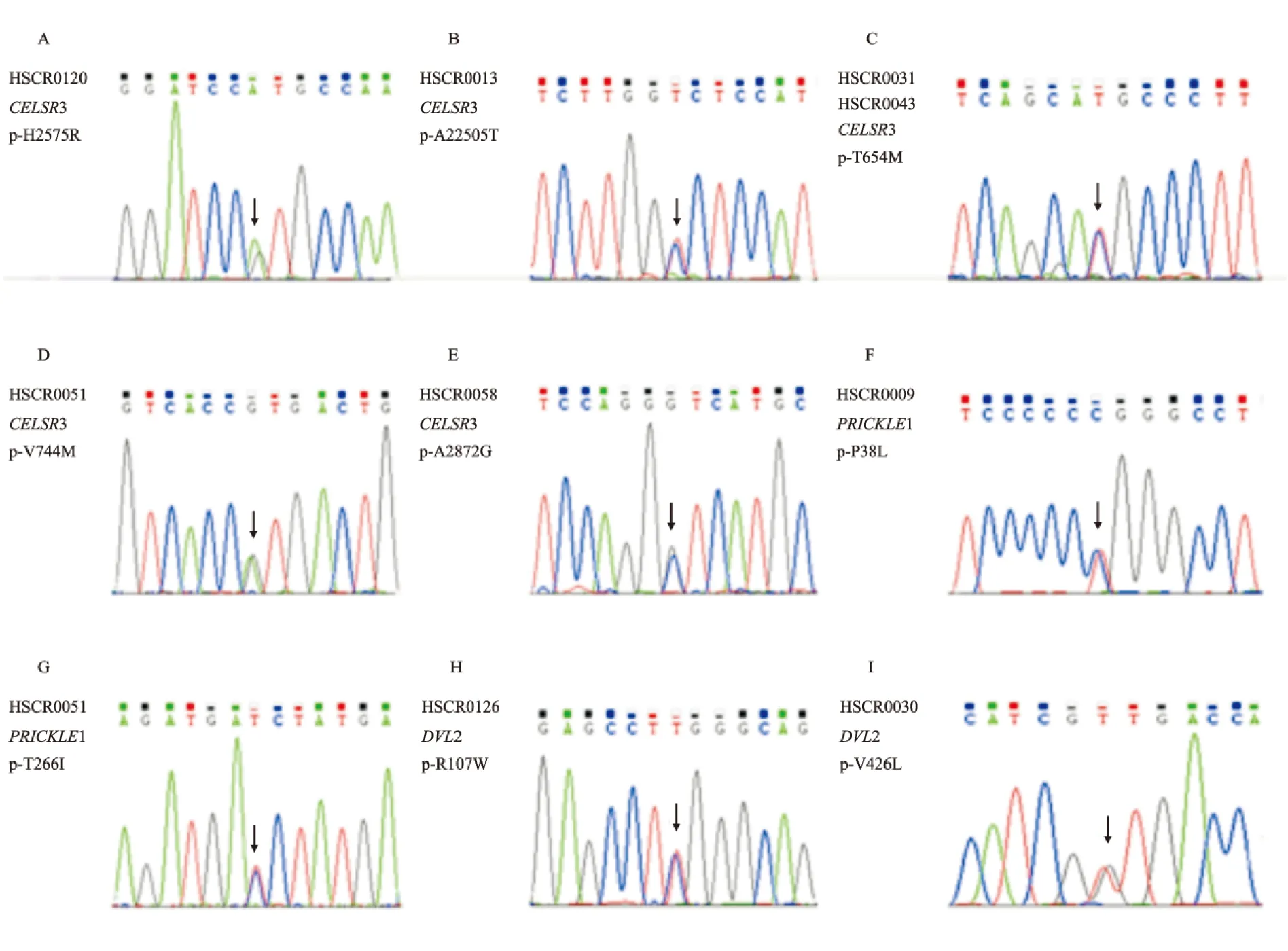
图1 CELSR3、PRICKLE1、DVL2的9个错义突变位点Sanger测序验证
A: HSCR0120患者CELSR3基因错义突变位点:c.7724A>G(H2575R);B:HSCR0013患者CELSR3基因错义突变位点:c.6613G>A(A2205T);C:HSCR0031和HSCR0048患者CELSR3基因错义突变位点:c.1961C>T(T654M);D:HSCR0051患者CELSR3基因错义突变位点:c.2230G>A(V744M);E:HSCR0058患者CELSR3基因错义突变位点:c.8615C>G(A2872G);F: HSCR0009患者PRICKLE1基因错义突变位点:c.113C>T(P38L);G:HSCR0051患者PRICKLE1基因错义突变位点:c.797C>T(T266I);H:HSCR0126患者DVL2基因错义突变位点:c.319C>T(R107W);I:HSCR0030患者DVL2基因错义突变位点:c.1276G>T(V426L)

图2 CELSR3基因编码蛋白模式图及本研究发现的5个错义突变位置
2.2 PCP通路核心基因突变患儿的临床表型 PCP通路核心基因突变共累及9例患儿,男7例,女2例,男性明显多于女性(3.5 ∶1),与HSCR发生的性别比接近。患儿临床分型分别为短段型4例,长段型3例,全结肠2例,长段型和全结肠型患儿的就诊年龄早于短段型。见表3。
3 讨论
HSCR是一种多基因、多因素参与的复杂性疾病,可散发,也可呈家族性遗传。目前已经鉴定的14个疾病易感基因主要来自ENS发育中起主要作用的3个信号通路(RET、EDNRB和SEMA3C/D)[2],以及可调控、影响这些基因表达的转录因子编码基因和其他几个在神经系统发育中已明确具有重要作用的基因。目前该病研究主要集中在与信号通路相关的肠神经嵴细胞发育以及肠道微环境等方面,关注神经网络形成、神经突起导向等方面的研究尚少。研究[7]表明PCP信号通路在中枢神经系统和外周神经系统的发育过程中如神经元迁移、轴突导向和树突形成等方面均起着不可或缺的作用。Celesr3和Fzd3基因敲除小鼠研究[6]也已证实PCP通路参与了小鼠肠神经系统的形成,推测PCP通路核心基因的功能异常可能与人类HSCR的易感性增高有关。本研究采用二代测序方法对83例患儿的PCP通路核心基因进行筛查,在9个患儿中发现了3个基因的9个杂合错义突变,其中6例存在CELSR3基因的有害突变,2例存在PRICKLE1基因的有害突变,2例存在DVL2的有害突变,1例患儿同时存在CELSR3基因和PRICKLE1基因的2个突变。全部9个错义突变累及的位点都具有高度保守性,平均GERP分值达到5.04(最低3.91,最高5.57),且在正常人群数据库中均未出现或者出现频率极低。
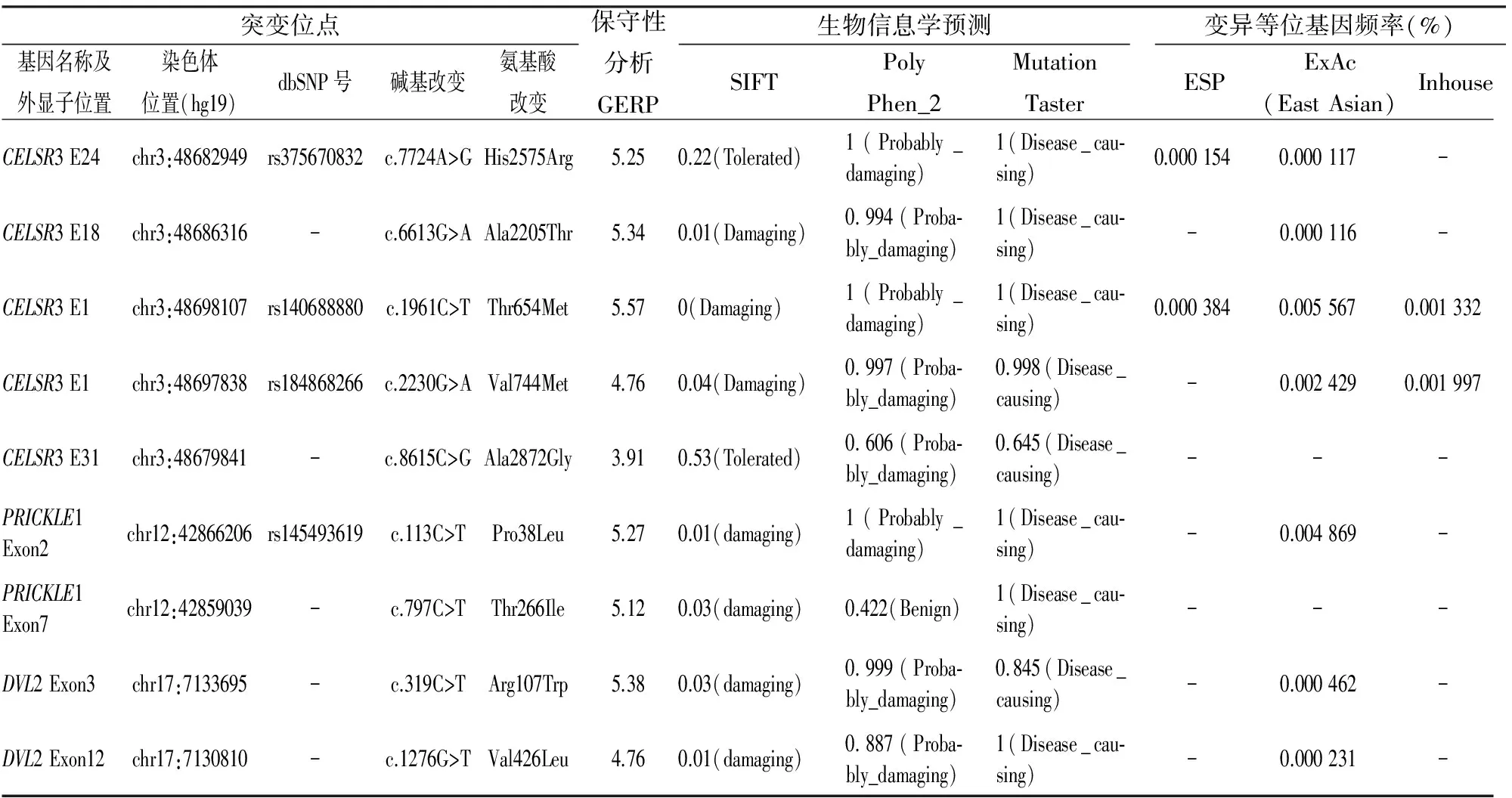
表2 HSCR患者PCP通路核心基因错义突变位点信息
表中的变异等位基因频率的数据分别来自ESP、ExAc和北京迈基诺基因科技有限责任公司自建数据库等3个正常对照人群数据库的数据
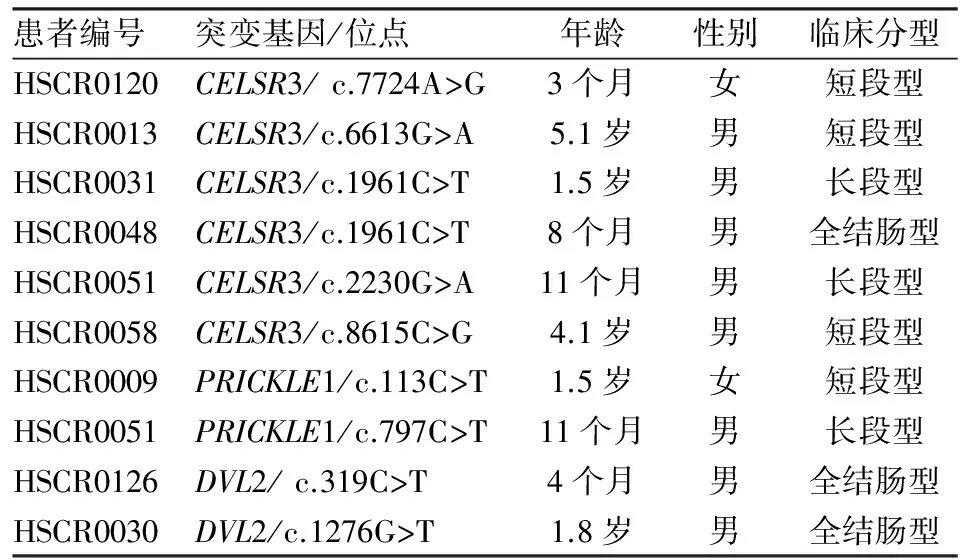
表3 PCP通路基因罕见突变累及患者的临床信息
CELSR3基因编码蛋白是长度超过3 000个氨基酸的非典型钙黏蛋白,其胞外域极大,其中8个N末端重复钙黏结构域(Cadherin)在细胞迁移中起重要作用[8]。神经嵴细胞是肠神经嵴细胞的祖细胞,其迁移障碍可导致其下游发生一系列变化,而CELSR3基因N端钙黏结构域可激活PCP通路并导致细胞极性变化,一旦发生错义突变可影响其蛋白功能,进而干扰其调控神经嵴细胞迁移功能[9]。CELSR3-/-小鼠模型研究[6]证实,突变小鼠病变段结肠呈现神经元迁移、轴突导向异常等多种障碍,其肠道运动功能的改变、排便障碍都和人类HSCR患者的临床表现高度相似。本研究CELSR3基因5个突变位点中有2个位于N末端重复钙黏结构域内,HSCR0031和HSCR0048患儿均存在编码区1961位“C”、“T”碱基互换,导致其编码的氨基酸由苏氨酸变成蛋氨酸,患儿表现为长段型巨结肠;HSCR0051患儿编码区2230位“G”、“T” 碱基互换,导致其编码的氨基酸由缬氨酸变成蛋氨酸(图2),患儿表现为全结肠型巨结肠,3例患儿病情均较严重。
作为PCP通路的核心基因,PRICKLE1基因编码的蛋白由831个 氨基酸构成,主要参与神经突的生长和神经元的迁移[10]。HSCR0009患儿编码区113位“C”、“T”碱基互换,导致其编码的氨基酸由脯氨酸变成亮氨酸,HSCR0051患儿编码区797位“C”、“T”碱基互换,导致其编码的氨基酸由苏氨酸变成异亮氨酸。DVL2同样是PCP通路的核心基因,编码蛋白由 736个氨基酸构成,HSCR0126患儿编码区319位“C”、“T”碱基互换,导致其编码的氨基酸由精氨酸变成色氨酸;HSCR0030患儿编码区1276位“G”、“T”碱基互换,导致其编码的氨基酸由缬氨酸变成亮氨酸。由于PCP通路的核心基因间相互紧密关联,任何一个基因的缺失或者功能受损都将影响其它核心蛋白的功能[7],因此PRICKLE1和DVL2基因的错义突变可能会干扰PCP通路在ENS发育中的正常作用,参与HSCR的发生。
综上所述,本研究通过对83例HSCR患儿PCP通路核心基因突变筛查,在9例患儿中检出3个核心基因的9个杂合突变位点,其编码蛋白的受累氨基酸均高度保守且在正常人群中发生率极低,提示PCP通路核心基因编码蛋白的功能异常可能是HSCR发病的原因之一,或可显著增加其发病的易感性。鉴于本研究的病例样本数有限,尚不能得出确切结论,有待于进一步增加样本量,并从蛋白表达、信号转导功能等多方面进行深入研究,以揭示人类ENS发育和HSCR发生的确切机制。
[1] Heanue T A, Pachnis V. Enteric nervous system development and Hirschsprung's disease: advances in genetic and stem cell studies[J]. Nat Rev Neurosci, 2007,8(6):466-79.
[2] Jiang Q, Arnold S, Heanue T, et al. Functional loss of semaphorin 3C and/or semaphorin 3D and their epistatic interaction with ret are critical to Hirschsprung disease liability[J]. Am J Hum Genet, 2015,96(4):581-96.
[3] Kapoor A, Jiang Q, Chatterjee S, et al. Population variation in total genetic risk of Hirschsprung disease from common RET, SEMA3 and NRG1 susceptibility polymorphisms[J]. Hum Mol Genet, 2015,24(10):2997-3003.
[4] Eichler E E, Flint J, Gibson G, et al. Missing heritability and strategies for finding the underlying causes of complex disease[J]. Nat Rev Genet, 2010,11(6):446-50.
[5] Tissir F, Goffinet A M. Shaping the nervous system: role of the core planar cell polarity genes[J]. Nat Rev Neurosci, 2013,14(8):525-35.
[6] Sasselli V, Boesmans W, Vanden Berghe P, et al. Planar cell polarity genes control the connectivity of enteric neurons[J]. J Clin Invest, 2013,123(4):1763-72.
[7] Goodrich L V. The plane facts of PCP in the CNS[J]. Neuron,2008,60(1):9-16.
[8] 曲宜波, 周立兵. 非典型钙粘蛋白Celsr1-3在神经系统发育和PCP中的功能[J]. 暨南大学学报(自然科学与医学版), 2012,33(4):344-50.
[9] Obermayr F, Hotta R, Enomoto H, et al. Development and developmental disorders of the enteric nervous system[J]. Nat Rev Gastroenterol Hepatol,2013,10(1):43-57.
[10]Yang T,Bassuk A G,Stricker S, et al. Prickle1 is necessary for the caudal migration of murine facial[J]. Cell Tissue Res, 2014,357(3):549-61.
网络出版时间:2016-8-10 11:04:49 网络出版地址:http://www.cnki.net/kcms/detail/34.1065.R.20160810.1104.032.html
Mutation screening of the core genes in planar cell polarity pathway in patients with Hirschsprung disease
Su Lin1, Jiang Qian2, Yu Kaihui3, et al
(1ReproductiveMedicineCenter,TheClinicalCollegeofPLAAffiliatedAnhuiMedicalUniversity,Hefei230031;2DeptofMedicalGenetics,BeijingMunicipalKeyLaboratoryofChildDevelopmentandNutriomics,CapitalInstituteofPediatrics,Beijing100020;3DeptofPathophysiology,SchoolofPreclinicalSciences,GuangxiMedicalUniversity,Nanning530021)
ObjectiveToinvestigatetheeffectofcoregenesmutationinplanarcellpolarity(PCP)pathwayonthepathogenesisofHirschsprungdisease(HSCR).Methods83ChinesechildrenwithHSCRwererecruitedinthisstudyafterobtaininginformedconsents,andgenomicDNAofallsubjectswasextractedfromperipheralblood.Thesuspectedmutationsof8coregenes(CELSR3,FZD3,VANGL1,VANGL2,PRICKLE1,PRICKLE2,DVL1,DVL2)inPCPpathwaywereverifiedbySangersequencingafterthenextgenerationsequencing.TherawdatawereanalyzedwiththemolecularbiologicalwebsitesandtheGeneToolsoftware.ResultsHeterozygousmissensemutationswere
Hirschsprungdisease;planarcellpolarity;genemutation; CELSR3
时间:2016-8-10 11:04:49
http://www.cnki.net/kcms/detail/34.1065.R.20160810.1104.031.html
2016-05-30接收
国家自然科学基金青年科学基金项目(编号:81300266);北京市自然科学基金面上项目(编号:7142029);北京市优秀人才培养个人项目(编号:2013D003034000007);北京市科技新星(编号:Z151100000315091)
1安徽医科大学解放军临床学院生殖中心,合肥 230031
2首都儿科研究所遗传研究室、儿童发育营养组学北京市重点实验室,北京 100020
3广西医科大学基础医学院病理生理学教研室,南宁 530021
首都儿科研究所附属儿童医院4新生儿外科、5病理科,北京 100020
苏 琳,女,硕士研究生;
姜 宏,男,教授,硕士生导师,责任作者,E-mail: jiangh105@sina.com
R 394.1;R 715.2;R 715.5;R 714.7;R 714.55
A
1000-1492(2016)10-1534-06
10.19405/j.cnki.issn1000-1492.2016.10.031
identifiedin9subjectswithadetectionrateof10.8%(9/ 83).Theywere: CELSR3genec.7724A>G(H2575R),c.6613G>A(A2205T),c.1961C>T(T654M),c.2230G>A(V744M)andc.8615C>G(A2872G); PRICKLE1genec.113C>T(P38L)andc.797C>T(T266I); DVL2genec.319C>T(R107W)andc.1276G>T(V426L),andallofthemhadnotbeenreportedbefore.Bioinformaticanalysisdemonstratedthatallthesemutationsweredeleteriousandmightbetheriskfactorsforpathogenesis.ConclusionAbnormalfunctionofPCPcoregenesmaybepossibleriskfactorforthepathogenesisofHSCR.
