CoⅡ(salen∗)存在下的氯丁二烯自由基聚合
2016-11-23李威威杨万泰付志峰
李威威,石 艳,杨万泰,付志峰
(北京化工大学化工资源有效利用国家重点实验室,北京100029)
CoⅡ(salen∗)存在下的氯丁二烯自由基聚合
李威威,石 艳,杨万泰,付志峰
(北京化工大学化工资源有效利用国家重点实验室,北京100029)
研究了N,N′⁃双(3,5⁃二叔丁基水杨醛)⁃1,2⁃环己二胺钴(Ⅱ)[CoⅡ(salen∗)]存在下氯丁二烯(CP)的自由基聚合,考察了不同溶剂、引发剂用量及配体对聚合反应的影响.结果表明,随着引发剂用量的增加,聚合反应的诱导期缩短,以[ABVN]0/[CoⅡ(salen∗)]0=3/1配比投料,聚合反应表现出较好的可控聚合特征.在苯、甲苯、四氢呋喃(THF)和乙酸乙酯(EA)4种溶剂中按照[CP]0/[CoⅡ(salen∗)]0/[ABVN]0=400/1/3的配比投料,在苯中的可控聚合程度最好:在低转化率(40%以下)实测聚合物分子量(MnGPC)与理论值(Mnth)吻合,且分子量随转化率增加呈线性增长.研究了THF、三乙胺(NEt3)、吡啶(Py)及水等不同配体对聚合反应的影响,发现在添加THF时,低转化率(40%以下)下Mn,GPC与Mn,th相符,分子量分布(PDI)相对较窄.
氯丁二烯;有机金属控制的自由基聚合;供电子配体;CoⅡ(salen∗);有机钴控制的自由基聚合
活性自由基聚合(LRP)已经发展成对聚合物的微结构、分子量以及分子量分布进行有效控制的重要手段.常见的LRP方法有氮氧稳定自由基聚合(NMRP)[1]、原子转移自由基聚合(ATRP)[2]和可逆加成断裂链转移(RAFT)自由基聚合[3].此外,还有其它一些LRP方法,如单电子转移自由基聚合[4]、碘化物存在下的自由基聚合[5]以及有机金属配合物控制的自由基聚合(OMRP)[6].一般认为OMRP的聚合过程同时存在可逆终止(RT)和退化链转移(DT)2种机理[7,8],反应式分别为
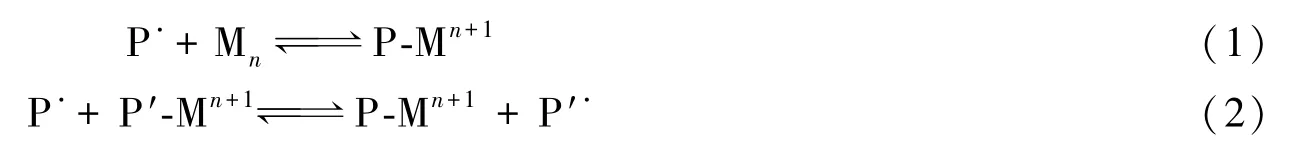
在OMRP中,所用的过渡金属有钛、铑、钒、钴等,其中钴配合物控制的自由基聚合的研究较多,统称为钴控制的自由基聚合(CMRP)[9].20世纪末,Wayland等[10]和Hardwood等[11]分别用有机钴卟啉和有机钴肟为引发剂实现了丙烯酸酯类单体的可控自由基聚合.10年后Jérôme等[12]以偶氮二甲氧基异庚腈(V⁃70)为引发剂在30℃下用乙酰丙酮钴[CoⅡ(acac)2]实现了乙酸乙烯酯(VAc)的可控聚合,成为LRP中一个了不起的突破.但研究发现CoⅡ(acac)2对丙烯酸酯类[12]和丙烯腈单体[13]并不适用,所得聚丙烯腈的Mn,GPC与Mn,th相差较大,且分子量分布(PDI在2.0左右)较宽.此外,用偶氮二异丁腈(AIBN)代替V⁃70时,CoⅡ(acac)2调控VAc的聚合反应[12]可控性则很差(PDI在2.0~3.5).最近,Peng等[14]以AIBN为引发剂在60℃下实现了CoⅡ(salen∗)对VAc和丙烯酸甲酯(MA)的聚合,并表现出较好的可控效果,说明CoⅡ(salen∗)对单体的适用范围优于CoⅡ(acac)2.
二烯类化合物是合成橡胶的重要单体,但关于其可控聚合的报道却很少.丁二烯和异戊二烯(Ip)通过NMRP[15,16](120~130℃)实现了可控自由基聚合;丁二烯的ATRP(110~130℃)可控效果很差[17,18];Ip的RAFT聚合研究结果差异很大[19,20].上述结果表明二烯类单体的可控聚合较困难. Ajellal等[21]研究了对Ip、氯丁二烯(CP)与含磷单体的稳定自由基共聚合(100℃),发现CP共聚合的可控程度很低,但Ip的共聚可控特征明显,说明CP的可控聚合相对更难.此外,工业化的二烯类橡胶是在高温(40~50℃)和低温(5~10℃)2种条件下生产的,因此更高温度下制备的二烯聚合产物也许
无实际价值.
本课题组通过RAFT聚合(60℃)[22]和碘代化合物调控的聚合(50℃,9℃)[23,24]实现了CP的可控聚合,并且在低温下制备的聚氯丁二烯具有优越的黏接性能.本文进一步研究了CoⅡ(salen∗)存在下CP的聚合,探讨钴配合物调控CP自由基聚合的可控性.研究了不同溶剂、配体以及引发剂用量对聚合可控程度的影响,与本体聚合进行了对比,并讨论了CoⅡ(salen∗)调控CP聚合的机理过程.
1 实验部分
1.1 试剂与仪器
CP,山西合成橡胶集团有限责任公司,蒸馏精制后使用;偶氮二异庚腈(ABVN,化学纯),武汉盛世精细化学品有限公司,用乙醇重结晶后使用;环己二胺(纯度98%)、乙酰丙酮钴(纯度97%),阿拉丁试剂公司;茴香醛(化学纯),天津市光复精细化工研究所;正戊烷、无水甲醇、无水乙醇、二氯甲烷、苯、甲苯、四氢呋喃(THF)和乙酸乙酯(EA)均为分析纯,北京市通广精细化工公司;经CaH2干燥处理精制后使用.
用日本TOSOH HLC⁃8320凝胶渗透色谱进行分子量和分子量多分散指数(PDI)测定.色谱柱为2根TSK gel Super Multipore HZ⁃M串联,淋洗液为THF,流速0.35 mL/min,测试温度40℃,用聚苯乙烯标样进行色谱柱校正;采用Brucker AV400核磁共振仪进行1H NMR测试,测试温度为25℃,以氘代氯仿(CDCl3)为溶剂,四甲基硅为内标;用Thermo Nexus 8700傅里叶变换红外光谱仪进行红外测试;紫外吸收采用UV⁃6100型紫外⁃可见分光光度计(上海美谱达仪器有限公司)测定.
1.2 实验过程
1.2.1 N,N′⁃双(3,5⁃二叔丁基水杨醛)⁃1,2⁃环己二胺(salen∗)的合成 在三口瓶中加入0.57 g 1,2⁃环己二胺和10 mL无水甲醇,搅拌下在0.5 h内将3,5⁃二叔丁基水杨醛的无水甲醇溶液(1.17 g,250μmol/mL)滴入三口瓶中,在氮气保护下加热回流6 h,析出黄色沉淀,抽滤,用无水甲醇洗涤3次,于50℃真空烘箱干燥,得到salen∗,产率约为68.8%;1H NMR(400 MHz,CDCl3),δ:13.63(s,2H),8.31(s,2H),7.31(s,2H),6.99 (s,2H),3.34(d,J=7.3 Hz,2H),1.94(d,J=12.8 Hz,2H),1.86(d,J=7.2 Hz,2H),1.73(d,J=9.4 Hz,2H),1.46(d,J=10.4 Hz,2H),1.40(s,18H),1.23(s,18H). salen∗的结构[25]如图1(A)所示.

Fig.1 Structures of salen∗(A)and CoⅡ(salen∗)(B)
1.2.2 钴配合物的合成 将CoⅡ(acac)2(113 mg,0.44mmol)和1.5mL CH2Cl2加入到单口瓶中,用高纯氩气鼓泡10 min,封口,抽真空、通氩气数次排除空气后,在氩气保护下将溶解在2.0 mL CH2Cl2中的salen∗(118 mg,0.44 mmol)注射入瓶中.室温下搅拌1 h,用40 mL的正戊烷沉淀CoⅡ(salen∗),离心干燥,产率约57.4%.IR(KBr压片),/cm-1:2951(vs),2866(s),1639(m),1595(s),1462 (m),1432(m),1386(m),1359(m),1338(m),1320(m),1254(s),1202(w),1175(s),929(m),869(m),835(m),786(m),746(w),640(w).CoⅡ(salen∗)的结构[26]如图1(B)所示.
1.2.3 实验方法 在装有搅拌子的50 mL单口瓶中顺次加入0.1637 g(0.27 mmol)CoⅡ(salen∗)、0.2017 g(0.81 mmol)ABVN和20 mL苯,用高纯氩气鼓泡10 min,封口,抽真空,通氩气数次排除空气后,用注射器将10mL氯丁二烯(CP)注入反应瓶中.将反应瓶置于50℃油浴中恒温反应,在预定时间取样,用称重法测定单体转化率.在上述反应的诱导期内,每隔30min取0.1mL的样品并用苯稀释至10 mL,进行紫外吸收测试.
2 结果与讨论
2.1 引发剂用量对反应的影响
OMRP的引发方式主要有2种:(1)以高氧化态的有机金属配合物Mtn+1⁃R[27]为引发剂和控制剂,
通过加热或光照的方式引发和调控聚合;(2)以较稳定的低价金属配合物与引发剂在加热或光照条件下原位生成Mtn+1⁃R[26]后引发和调控聚合.本文采用常用的第二种方式进行引发.
以ABVN为引发剂,苯为溶剂,在50℃下进行CP的自由基聚合.考察了引发剂用量对聚合和可控性的影响.
图2为不同引发剂用量下CP聚合反应动力学关系图.可以看出,反应均存在诱导期.诱导期是引发剂分解生成的自由基与二价钴反应形成三价钴所需的时间.不同引发剂与催化剂配比下的诱导期有明显的差异,引发剂量最少的[ABVN]0/[CoⅡ(salen∗)]0=2/1体系,诱导期约为 280 min,[ABVN]0/[CoⅡ(salen∗)]0=2.5/1体系诱导期约为220min,配比为3/1时诱导期约为120min,可见诱导期随引发剂用量增大在不断缩短.当配比大于3/1时诱导期基本稳定在90 min左右.诱导期结束后,在所用的5组配比下,ln([M]0/[M])与反应时间在考察范围内都呈线性关系,表明聚合体系是一级动力学关系,体系中活性中心的浓度基本恒定.图2中随引发剂用量增加直线的斜率逐渐变大,即聚合反应速率越快.聚合反应速率取决于体系中自由基的浓度[R·],引发剂用量越大聚合反应速率越快,说明CoⅡ(salen∗)调控下CP聚合的机理应为DT历程[7].另外,结合体系有明确的诱导期以及没有出现RT机理常见的二段聚合现象[28],也间接说明此调控机理是DT过程,这与文献[14]中所述VAc的调控机理一致,而与MA的(认为是RT的机理)不同[14].
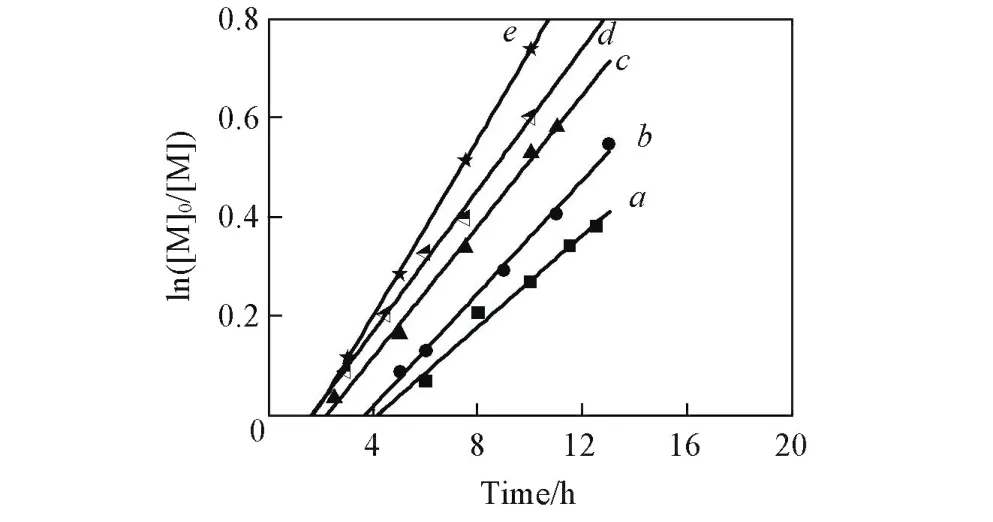
Fig.2 First order kinetic plots for polymerization of CP in benzene [V(CP)/V(benzene)=1/2,50℃]with different ABVN concentrations
图3是所得聚合物的数均分子量(Mn)、PDI与转化率关系图.可以看出在5组实验中,当[ABVN]0/[CoⅡ(salen∗)]0=3/1时,聚合反应表现出很好的可控聚合特征.在转化率低于40%时,聚合物Mn,GPC与Mn,th十分接近,且随单体转化率的增加基本呈线性增长,PDI=1.8左右;转化率高于40%,Mn,GPC逐渐低于Mn,th,且分子量增长不明显.其它比例下的聚合也均出现了随转化率增加分子量不再增长的现象,这种现象在CoⅡ(TMP)调控下VAc的聚合[29]与CoⅡ(ketoaminato)及其衍生物调控下MA的聚合(转化率上限约为 26%)[28]中都出现过.随着引发剂用量的略增,[ABVN]0/[CoⅡ(salen∗)]0为3.5/1~4/1时,Mn,GPC与 Mn,th偏差变大,聚合反应的可控程度有所下降.而当[ABVN]0/[CoⅡ(salen∗)]0小于3/1时,随着引发剂用量增加聚合反应的可控效果也逐渐变差.按照DT机理,当加入的热引发剂适度过量时能够有效地将CoⅡ转化为CoⅢ⁃R,且保证增长链自由基的浓度
适当,才会表现出较好的可控效果,因此存在最佳的引发剂与 CoⅡ的配比.当[ABVN]/[CoⅡ(salen∗)]小于3/1时,加入的引发剂的量较少可能不足以将所有的CoⅡ转化为CoⅢ⁃R[28],此时反应过程中可能同时包含了RT和DT 2种聚合机理,不适用于CP的聚合反应,致使可控效果不佳.高转化率下分子量不增长的原因有以下两个方面:一个是增长链自由基的β⁃氢转移反应,在高温下反应比较明显[30];另一个是CP等二烯类单体在较高转化率时容易发生支化副反应(CP聚合反应中有1,2加成、1,4加成和3,4⁃加成3种形式,其中1,2加成在分子链中生成的烯丙基氯以及3,4⁃加成生成的乙烯基侧基均是易支化的结构),而支化分子的流体力学体积要比相同分子量的线性分子小.另外,链自由基间不可逆双基终止的比例随转化率增大逐步累积(这一点与RAFT聚合体系一致).
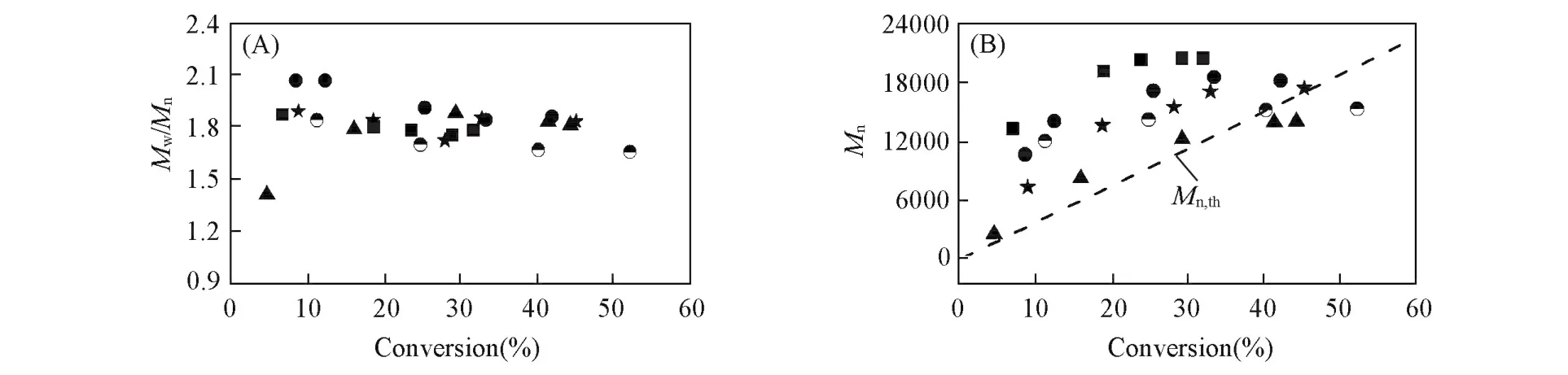
Fig.3 Molecular weight distribution(A)and number⁃averagemolecular weight(B)for CoⅡ(salen∗)me⁃diated CP polymerization in benzene(CP/benzene=1/2,50℃)with different ABVN concentrations
上述实验结果比Peng等[14]运用CoⅡ(salen∗)来调控MA的聚合效果稍差,但要比Kermagoret等[26]运用CoⅡ(salen∗)的衍生物来调控丙烯酸丁酯(nBA)(虽然转化率能达到60%,但前期40%以内Mn,GPC远高于Mn,th)与 Sherwood等[28]运用CoⅡ(ketoaminato)及其衍生物调控 MA(总的转化率低于30%,Mn,GPC远高于Mn,th)的可控程度要高得多.另外,虽然本文所得PCP的PDI数值较高,但对于橡胶的实际应用而言宽分布对加工性能是十分有利的[31].另外,本文中引发剂相对于CoⅡ的用量远低于文献[14,30]中常用的比例(一般为6/1~10/1),这可能与CP单体的高活性有关.
2.2 溶剂对聚合反应的影响
一般配位型和非配位型溶剂对钴调控聚合的可控程度有较大影响[29],本文选取苯、甲苯、四氢呋喃(THF)、乙酸乙酯(EA)4种溶剂进行溶液聚合,其中苯、甲苯、乙酸乙酯(EA)为非配位型溶剂,THF为配位型溶剂.
图4是CP本体聚合和在不同溶剂中的聚合反应动力学关系图.可以看出,ln[M]0/[M]与反应时间在考察范围内都呈线性关系,表明反应体系中活性中心的浓度基本保持恒定.相对于本体聚合,溶液聚合的诱导期均变长.所用的4种溶剂中,苯中的诱导期较短(120min),THF和EA中的诱导期相近(170min),甲苯中的诱导期则较长(270 min).不同的诱导期表明了溶剂效应对引发剂的分解、二价钴和自由基的反应均有一定的影响.此外,从图4可以看出,4种溶剂中的聚合速率有所不同,苯和甲苯中的速率接近,THF和EA中的略快.这些结果与Peng等[29]报道的溶剂对VAc聚合的影响不同:配位型溶剂THF可缩短诱导期,而非配位型溶剂如苯中的诱导期则较长且接近.

Fig.4 First order kinetic plots for polymerization of CP in different solutions at 50℃
图5(A)和(B)是所得聚合物的Mn和PDI与转化率的关系图.可以看出,在本体聚合中,低转化率下聚合物分子量随转化率增加而增大,但Mn,GPC与Mn,th的差距也随转化率不断增大.当单体转化率高于30%后,分子量不再增长.与本体聚合相比,以苯为溶剂时可控效果明显提高,在低转化率时,Mn,GPC与Mn,th十分接近,转化率达40%左右后分子量不再有明显的增长.其它3种溶剂中,也均出现了单体转化率达一定值后分子量不增长的现象.以THF、甲苯、EA为溶剂时,分子量增长的转化率上限大约分别为40%,30%和30%.综合对分子量及PDI的控制,以甲苯为溶剂最好,其次为THF.EA中的分布较窄,但Mn,GPC与Mn,th相差较大.图5(C)是以苯为溶剂所得聚合物的凝胶色谱(GPC)图可见,曲线都成单峰分布,峰型基本对称且随转化率增加分子量逐渐增大,本体聚合以及在THF中聚合物的GPC曲线与图5(C)类似.
2.3 添加配体对聚合反应的影响
添加配体可显著改变CMRP的可控效果[32,33].因此,本文进一步研究了配体对CP自由基聚合的影响.考察了THF、三乙胺(NEt3)、吡啶(Py)、水及二甲基亚砜(DMSO)5种电子配体(ED)对聚合的影响.反应条件:苯为溶剂,[CP]0∶[ABVN]0∶[CoⅡ(salen∗)]0=400∶3∶1,[Co]∶[ED]=1∶25.
从图6的反应动力学关系图中可以看出,添加配体使得体系的诱导期均有所缩短,这一发现与Peng等[29]和Matyjaszewski等[32]研究的配体对VAc聚合反应动力学的影响一致.添加THF的诱导期最短,约为60min.添加水的动力学曲线与Py的动力学曲线基本吻合(图中没给出).从图6还可看出,添加THF的聚合速率与空白样基本一致,而其它的3个体系聚合速率均有提高.
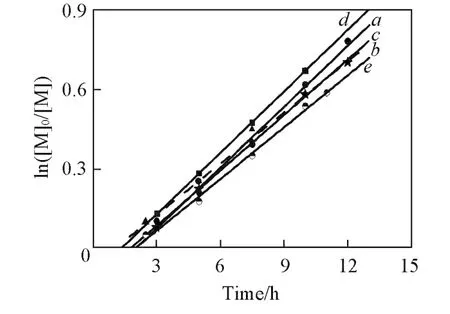
Fig.6 First order kinetic p lots for polymerization of CP in benzene at 50℃
图7是所得聚合物的Mn,PDI与转化率关系以及GPC曲线图.可以看出,添加水的体系中Mn,GPC远高于Mn,th,且分子量基本不增长.当添加Py,NEt3时,虽然聚合中分子量均有明显的增长,但与不添加配体的样品相比,Mn,GPC偏高于Mn,th,且分子量分布较宽(PDI均在2.0左右).添加DMSO后,低转化率下Mn,GPC与Mn,th更吻合,但PDI变大(1.95~2.21).与不添加配体的样品相比,添加THF时在0~40%的转化率区间内表现出更好的可控效果,Mn,GPC与Mn,th吻合,PDI稳定在(1.6~1.8)之间.图7(C)是添加THF所得聚合物的GPC曲线叠加图,可以看出曲线都成单峰分布,且随转化率增加,聚合物分子量逐渐增加.

Fig.7 Plots ofmolecular weight(A)and PDI(B)versus conversion for the addition of electron donors to polymerization of CP in benzene at 50℃and typical GPC traces from the synthesis of PCP with addition of THF(C)
2.4 CoⅡ(salen∗)和CoⅢ(salen∗)⁃R的紫外吸收
图8是在[CP]0∶[ABVN]0∶[CoⅡ(salen∗)]0=400∶3∶1条件下,诱导期内体系的紫外吸收与时间的关系图.可以看出,随着反应时间的延长,360 和419 nm处吸收峰的强度在不断减弱,反应到120 min时仅在410 nm处有1个较强的吸收峰.这一结果与Ren等[34]的报道一致:CoⅡ(salen∗)在360和430 nm有强吸收峰,而CoⅢ(salen∗)⁃R仅在410 nm左右有吸收峰.这表明在诱导期内CoⅡ(salen∗)与引发剂生成的自由基反应不断地向CoⅢ(salen∗)⁃R转化,反应至120min时(即诱导期结束时),CoⅡ(salen∗)的特征峰消失,只有410 nm处的吸收峰,说明CoⅡ(salen∗)已经完全转化为CoⅢ(salen∗)⁃R,之后的聚合应该是按照DT机理调控CP的聚合过程.
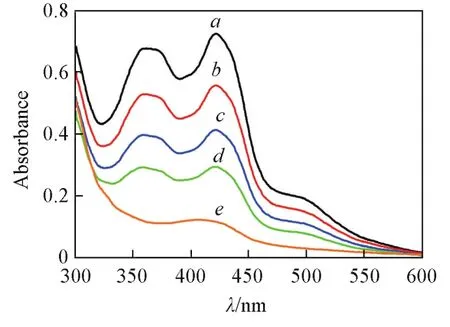
Fig.8 Time dependent UV⁃Vis spectra illustrating the transformation of CoⅡ(salen∗) to CoⅢ(salen∗)⁃R during the induction period of CP polymerization
此外,由于钴的顺磁性,PCP⁃CoⅡ(salen∗)核磁共振氢谱发生严重变形,与RAFT聚合或传统自由基聚合所得的 PCP谱图完全不同,不能反映PCP的真实结构和信息,因此对聚合物的结构没有做进一步的分析,但也间接证实了 PCP⁃CoⅡ(salen∗)结构的存在.
3 结 论
研究了CoⅡ(salen∗)调控的氯丁二烯自由基聚合.考察了不同溶剂对聚合反应的影响,发现苯中低转化率下实测分子量与理论值符合的最好;苯中的反应诱导期较短;苯溶液中的聚合可控程度明显优于本体聚合.随着引发剂用量增加,反应的诱导期逐渐缩短,以[ABVN]0/[CoⅡ(salen∗)]0=3/1投料聚合反应表现出较好的可控聚合特征.反应动力学和紫外吸收结果分析表明,聚合的机理为DT历程.添加Py和NEt3配体时,聚合的可控程度变差;添加THF时表现出最好的可控效果.
[1] Georges M.K.,Veregin R.P.N.,Kazmaier P.M.,Hamer G.K.,Macromolecules,1993,26(11),2987—2988
[2] Matyjaszewski K.,Tsarevsky N.V.,J.Am.Chem.Soc.,2014,136(18),6513—6533
[3] Goto A.,Sato K.,Tsujii Y.,Fukuda T.,Moad G.,Rizzardo E.,Thang S.H.,Macromolecules,2001,34(3),402—408
[4] Lligadas G.,Ladislaw J.S.,Guliashvili T.,Percec V.,Journal ofPolymer Science Part A:Polymer Chemistry,2008,46(1),278—288
[5] Iovu M.C.,Matyjaszewski K.,Macromolecules,2003,36(25),9346—9354
[6] Peng C.H.,Yang T.Y.,Zhao Y.,Fu X.F.,Org.Biomol.Chem.,2014,12(43),8580—8587
[7] Peng C.H.,Li S.,Wayland B.B.,Journal of the Chinese Chemical Society,2009,56(2),219—233
[8] Peng C.H.,Li S.,Wayland B.B.,ACSSymposium Series,2009,1024,115—129
[9] Zhao Y.G.,Yu M.M.,Liu Y.C.,Fu X.F.,Scientia Sinics Chimica,2014,44(2),236—253(赵亚光,禹蒙蒙,刘禹初,付雪峰.中国科学:化学,2014,44(2),236—253)
[10] Wayland B.B.,Poszmik G.,Mukerjee S.L.,Journal of the American Chemical Society,1994,116(17),7943—7944
[11] Arvanitopoulos L.D.,Greuel M.P.,Harwood H.J.,Polym.Prepr.Am.Chem.Soc.,1994,35,549—550
[12] Debuigne A.,Caille J.,Jérôme R.,Angewandte Chemie International Edition,2005,44(7),1101—1104
[13] Buchmeiser M.R.,Marino M.G.,MacromolecularMaterialsand Engineering,2012,297(9),894—901
[14] Peng C.H.,Liao C.,Hsu C.,Wang F.,Wayland B.B.,Polymer Chemistry,2013,4(10),3098—3104
[15] Benoit D.,Harth E.,Fox P.,Waymouth R.M.,Hawker C.J.,Macromolecules,2000,33(2),363—370
[16] Georges M.K.,Hamer G.K.,Listigovers N.A.,Macromolecules,1998,31(25),9087—9089
[17] Hua J.,Li X.,Li Y.S.,Xu L.,Li Y.X.,Journal ofApplied Polymer Science,2007,104(6),3517—3522
[18] Yang H.Q.,Hua J.,Xu L.,Huang B.C.,China Synthetic Rubber Industry,2006,29(3),228(杨海强,华静,徐玲,黄宝琛.合成
橡胶与工业,2006,29(3),228)
[19] Jitchum V.,Perrier S.,Macromolecules,2007,40(5),1408—1412
[20] Germack D.S.,Wooley K.L.,Journal ofPolymer Science Part A:Polymer Chemistry,2007,45(17),4100—4108
[21] Ajellal N.,Thomas C.M.,Carpentier J.,Polymer,2008,49(20),4344—4349
[22] Hui J.,Dong Z.J.,Shi Y.,Fu Z.F.,YangW.T.,RSC Adv.,2014,4(98),55529—55538
[23] Hui J.,Shi Y.,Li T.,Fu Z.F.,Yang W.T.,RSC Adv.,2015,5(55),44326—44335
[24] Gu J.M.,Fu Z.F.,Yang W.T.,Shi Y.,Journal of Applied Polymer Science,2013,128(4),2291—2296
[25] Shi Y.Q.,Synthesisand Catalytic Activity ofSchiffBase CopperComplexes,Zhejiang Sci⁃Tech University,Hangzhou,2013(时永强.席夫碱铜配合物的合成及催化性能的研究.杭州:浙江理工大学,2013)
[26] Kermagoret A.,Jérôme C.,Detrembleur C.,Debuigne A.,European Polymer Journal,2015,62,312—321
[27] Zhao Y.,Yu M.,Fu X.,Chemical Communications,2013,49(45),5186—5188
[28] Sherwood R.K.,Kent C.L.,Patrick B.O.,McNeilW.S.,Chemical Communications,2010,46(14),2456—2458
[29] Hsu C.,Yang T.,Peng C.,Polym.Chem.,2014,5(12),3867—3875
[30] Lin Y.,Hsieh Y.,Lin Y.,Peng C.,Macromolecules,2014,47(21),7362—7369
[31] Zhang J.G.,Wen Q.,Wang Y.J.,Zhang X.J.,World Rubber Industry,2013,40(07),28—35(张建国,文强,王永军,张新军.世界橡胶工业,2013,40(07),28—35)
[32] Maria S.,Kaneyoshi H.,Matyjaszewski K.,Poli R.,Chemistry:A European Journal,2007,13(9),2480—2492
[33] Debuigne A.,Champouret Y.,Jérôme R.,Poli R.,Detrembleur C.,Chemistry:A European Journal,2008,14(13),4046—4059
[34] Ren W.,Wang Y.,Zhang R.,Jiang J.,Lu X.,Journal ofOrganic Chemistry,2013,78(10),4801—4810
Cobalt⁃mediated Radical Polymerization(CMRP)of Chloroprene by CoⅡ(salen∗)†
LIWeiwei,SHIYan∗,YANGWantai,FU Zhifeng
(State Key Laboratory ofChemical Resource and Engineering,Beijing University ofChemical Technology,Beijing 100029,China)
Radical polymerization of chloroprene(CP)in the presence of N,N′⁃bis(3,5⁃di⁃tert⁃butylsalicyli⁃dene)⁃1,2⁃cyclohexanediamine[CoⅡ(salen∗)],as amediator was investigated.The results show that the induction time is shortened and the polymerization rate is increased with the increase of ABVN concentrations. The linear increase inmolecular weightwith conversion,and molecular weight close to the theoretical value at low conversion in benzene,demonstrating the living process of CP radical polymerization mediated by [CoⅡ(salen∗)].In order to determinewhat effect,if any,the solvents had upon the control of the polymeri⁃zation,various solventswere used in the CMRP of chloroprene,and the process in benzene efficiently controls themolarmass(characterized by size exclusion chromatography,SEC)at low monomer conversion.Then,the effect of electron donors(ED)on the[CoⅡ(Salen∗)]⁃mediated radical polymerization of CP was examined by the addition of electron donors,water,pyridine(Py),NEt3,tetrahydrofuran(THF)and dimethylsulfoxide (DMSO)to the complex in the molar ratio[CP]/[Co]/[ABVN]/[ED]=400/1/3/25.The induction periods became shorter with the addition of electron donors(ED).Obviously,the control of polymerization increased with the addition of THF,which was demonstrated bymolecularweight close to the theoretical value and a relatively narrow molecular weight distribution at low monomer conversion.
Chloroprene;Organometallic mediated radical polymerization;Electron donor;CoⅡ(salen∗);Cobalt⁃mediated radical polymerization
O631
A
10.7503/cjcu20160496
(Ed.:D,Z)
†Supported by the National Natural Science Foundation of China(No.51373021).
2016⁃07⁃13.
日期:2016⁃10⁃14.
国家自然科学基金(批准号:51373021)资助.
联系人简介:石 艳,女,博士,副教授,博士生导师,主要从事可控自由基聚合和乳液聚合.E⁃mail:shiyan@mail.buct.edu.cn
