4,7-二氮杂螺[2.5]辛烷-7-甲酸叔丁酯的合成
2016-11-19冯瑾瑾王慧东陈楚珺刘宝贵黄齐茂
冯瑾瑾, 朱 慧, 王慧东, 陈楚珺, 刘宝贵, 黄齐茂*
(1. 武汉工程大学 化工与制药学院,湖北 武汉 43007; 2. 武汉市普仁医院,湖北 武汉 430081)
·制药技术·
4,7-二氮杂螺[2.5]辛烷-7-甲酸叔丁酯的合成
冯瑾瑾1, 朱 慧2, 王慧东1, 陈楚珺1, 刘宝贵1, 黄齐茂1*
(1. 武汉工程大学 化工与制药学院,湖北 武汉 43007; 2. 武汉市普仁医院,湖北 武汉 430081)
以丙二酸二乙酯为原料,通过环合、霍夫曼反应、水解、酰化和还原等反应合成了医药中间体4,7-二氮杂螺[2.5]辛烷-7-甲酸叔丁酯,总收率9.16%,其结构经1H NMR和MS确证。
丙二酸二乙酯; 4,7-二氮杂螺[2.5]辛烷-7-甲酸叔丁酯; 霍夫曼反应; 药物合成
医药中间体一般都是高附加值[1]的精细化工产品,开发这一系列产品,是企业寻求新的经济增长点和主要出路,市场广阔、生命力持久[2-4]。在亚洲,有机医药中间体生产国中,我国也是最具有市场竞争力的国家之一,医药中间体约360个,出口220个,年产量约2.5万吨,每年出口约1.4万吨[5-6]。农药、医药及其中间体工业是精细化工领域的重要门类,制药工业的发展速度在所有工业中是比较突出的[7-10]。
4,7-二氮杂螺[2.5]辛烷-7-甲酸叔丁酯(9)是一种重要的医药中间体,是很多药物及辅助剂[11]的重要合成原料。用于酪氨酸激酶及一些蛋白激酶抑制剂等的制备。其衍生品可作为一种蛋白酶抑制剂和抗肿瘤剂[12]。酪氨酸激酶抑制剂阻断酪氨酸激酶的活性,抑制细胞增殖,已开发多种抗肿瘤药物。蛋白激酶抑制剂及其制备药物成分可治疗心力衰竭和癌症,尤其是JAK激酶抑制剂[13-14],以及一些抗病毒药物和抗高血压药物等,阻断信号转导介导肿瘤细胞mRNA的表达,诱导肿瘤细胞凋亡。9的合成路线主要有两种。路线一:以1-苄基氧羰基氨基-1-环丙烷甲酸为起始原料,在碱性条件下与氨乙酸乙酯加成,经钯催化还原,再回流关环,经三氟化硼乙醚和硼氢化钠进一步还原后与二碳酸二叔丁酯加成得9。该方法对设备要求高且总收率较低。路线二:以氯乙酸乙酯和乙二胺为起始原料,与乙醇钠加成环合得2-吡嗪酮,再与三乙胺和二碳酸二叔丁酯加成,经氢化钠催化与苄溴加成,在格式试剂和异丙醇钛作用下于-78 ℃回流,再和氯化铵反应,继而经钯碳还原得9。该方法总合成率较低,反应条件存在安全隐患。以上方法均存在以下缺点:(1)原料昂贵、成本高;(2)操作复杂,收率低;(3)条件苛刻,对设备要求高;(4)常规环境下,原料性质不稳定,合成路线存在安全隐患。
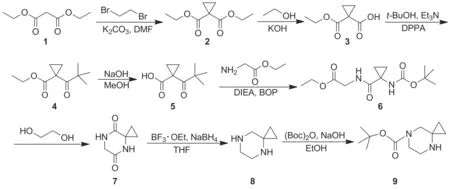
Scheme 1
本文针对以上问题设计了一种合成9的新方法[15-22]。以丙二酸二乙酯(1)为起始原料,经环合反应制得1,1-环丙烷二甲酸二乙酯(2); 2经水解制得1,1-环丙烷二甲酸单乙酯(3); 3经霍夫曼反应重排得1-叔丁氧羰基氨基-1-环丙烷甲酸乙酯(4); 4水解得1-叔丁氧羰基氨基-1-环丙烷甲酸(5); 5经α-氨基乙酸乙酯酰化得1-叔丁氧羰基氨基-1-环丙烷羰基氨基乙酸乙酯(6); 6用乙二醇再次环合得5,8二酮-4,7-二氮杂螺[2.5]辛烷(7); 7经还原反应得4,7-二氮杂螺[2.5]辛烷(8); 8与二碳酸二叔丁酯经加成反应合成9,总收率9.16%,其结构经1H NMR和MS确证。
该合成方法具有工艺简单、原料廉价易得、反应条件温和并较易控制、成本低及产率高等优点。
1 实验部分
1.1 仪器与试剂
Agilent 400MR型核磁共振仪[(CD3)2SO为溶剂,TMS为内标];LTQXL型高效液相色谱-线性离子阱质谱联用仪。
所用试剂均为化学纯或分析纯。
1.2 合成
(1) 2的合成
在三口瓶中依次加入1 28.82 g(0.18 mol), 1,2-二溴乙烷40.56 g(0.22 mol),碳酸钾62.10 g(0.45 mol),四丁基溴化铵0.17 g(5.27×10-4mol)和DMF 150 mL,搅拌下回流(80 ℃)反应15~16 h;于100 ℃反应2~3 h。冷却至室温,过滤,滤液蒸除溶剂得无色油状液体2 25.53 g,产率87.2%。
(2) 3的合成
在三口瓶中加入2 25.53 g(0.14 mol)和乙醇84 mL,搅拌下分批加入氢氧化钾7.84 g(0.14 mol,控制温度<30 ℃),加毕,于室温反应过夜。旋蒸脱溶,加入适量水,用混合溶剂[A=V(石油醚) ∶V(乙酸乙酯)=1 ∶1)]萃取,水相用浓盐酸调至pH 2,用乙酸乙酯萃取,合并萃取液,旋蒸脱溶得黄色油状液体3 20.17 g,产率93.0%。
(3) 4的合成
在三口瓶中依次加入3 20.17g(0.13 mol),t-BuOH 145 mL, 叠氮磷酸二苯酯(DPPA)35.13 g(0.13 mol)和三乙醇胺16.76 g(0.17 mol),搅拌下于室温反应2~4 h;回流反应3~4 h。旋蒸脱溶,用混合溶剂[V(石油醚) ∶V(乙酸乙酯) ∶V(饱和Na2CO3溶液)=1 ∶1 ∶1]萃取,萃取液旋蒸脱溶得白色固体4 25.93 g,产率88.7%。
(4) 5的合成
在三口瓶中依次加入4 25.93 g(0.11 mol), NaOH 9.05 g(0.22 mol)和乙醇48 mL,搅拌下于室温反应过夜。旋蒸脱溶,用乙醇洗涤,用浓盐酸调至pH 3(析出大量固体),过滤,滤饼干燥得白色结晶5 19.57 g,产率86.0%;1H NMRδ: 0.951(s, 2H, CH2), 1.262(s, 2H, CH2), 1.374(s, 9H, CH3), 2.509(s, 1H, COOH), 3.376(s, 1H, NH)。
(5) 6的合成
将5 19.57 g(0.10 mol),α-氨基乙酸乙酯13.59 g(0.10 mol),N,N-二异丙基乙胺(DIEA) 31.40 g(0.25 mol)和六氟磷酸苯并三唑-1-氧基三(二甲氨基)磷(BOP)51.47 g(0.12 mol)混合,加入二氯甲烷250 mL,搅拌下于室温反应过夜。依次用1 mol·L-1盐酸(10 mL)和饱和Na2CO3水溶液(10 mL)洗涤,旋蒸脱溶,经硅胶柱层析(洗脱剂:A=10 ∶1)纯化得乳白色固体6 5.30 g,产率19.0%;1H NMRδ: 0.900(s, 2H, CH2), 1.171(s, 2H, CH2), 1.171(m, 3H, CH2CH3), 1.392(s, 9H, CH3), 2.519(s, 1H, CONH), 3.338(s, 1H, OCONH), 3.811(s, 2H, OOCNH), 4.109(s, 2H, NCH2CO)。
(6) 7的合成
在三口瓶中依次加入6 5.3 g(0.02 mol)和乙二醇34 mL,搅拌下于150 ℃反应过夜;回流反应至完全(TLC跟踪)。减压蒸除溶剂,加入乙醇5 mL,过滤,滤饼干燥得乳白色固体7 2.25 g,产率86.7%;1H NMRδ: 2.06(s, 2H, CH2), 2.00(s, 2H, CH2), 3.858(s, 2H, NCH2), 8.028(s, 1H, NHCO), 8.272(s, 1H, NHCO); MSm/z: 140.28[M+]。
(7) 8的合成
N2保护下,在三口瓶将7 2.25 g(0.02 mol),三氟化硼乙醚11.4 g(0.10 mol)和硼氢化钠3.05 g(0.10 mol)混合,加入THF 85 mL,回流(66 ℃)反应过夜。控制温度为5 ℃,逐滴加入适量乙醇10 mL,蒸馏除去乙醇和THF,用二氯甲烷/乙醇(V/V=1/1)重结晶,过滤,滤饼干燥得棕色固体8 1.5 g,产率93.8%。
(8) 9的合成
在三口瓶中将8 1.5 g(0.01 mol), (Boc)2O 5.84 g(0.02 mol)和NaOH 1.18 g(0.04 mol)混合,搅拌下缓慢滴加乙醇16 mL(<5 ℃),滴毕,缓慢升至室温,反应过夜。过滤,滤液旋蒸脱溶后经硅胶柱层析(梯度洗脱剂:A=10 ∶1~3 ∶1)纯化得9 2.72 g,产率95.7%,总收率9.16%;1H NMR(CDCl3)δ: 0.564(s, 4H, CH2), 1.729(s, 9H, CH3), 2.883(s, 2H, CH2), 3.234(s, 2H, CH2), 3.382(s, 2H, CH2), 4.115(s, 1H, NH); MSm/z: 212.46[M+]。
2 结果与讨论
2.1 合成
(1) 2的合成
在2的合成中,温度决定反应速率,但是在较高温度下反应,实验过程中所放出的气体量较大而冲料;温度较低则反应进行不彻底而影响收率。在保证其他条件不变的情况下,改变回流温度分别为120 ℃, 140 ℃, 160 ℃和180 ℃,考察其对反应的影响,结果见表1。由表1可知,不同反应温度,收率相差较大。在120 ℃反应时,反应达不到此过程需要一定的活化能,原料转化率低,从而使环合反应进行程度不同。反应温度在180 ℃时,副产物增加。因此,确定最适宜的反应温度为160 ℃。
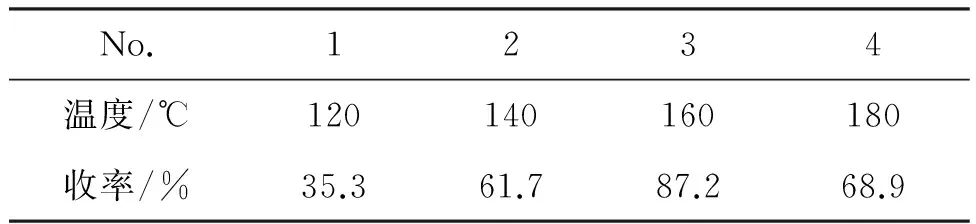
表1 反应温度对环合反应的影响
(2) 4的合成
在4的合成中,后处理中溶剂的选择对反应的收率影响较大。考察了不同萃取溶剂对4收率的影响,结果见表2。由表2可见,选择溶剂为PE ∶EA=1 ∶1(V∶V)的混合溶剂,可以成功提高反应的收率。后处理蒸馏除去叔丁醇,加入EA和饱和碳酸钠1∶1的混合溶液洗涤,分层很不明显。加入等量的PE,可迅速分出有机相。因此,后处理萃取溶剂应选择PE ∶EA=1 ∶1,可使收率显著提高。

表2 萃取溶剂对霍夫曼反应收率的影响
(3) 6的合成
BOP试剂常用在加成缩合反应中,与有机碱DIEA同用。选择合适的投料量,可以提高反应的收率。因此对BOP的投料量进行了对比,结果见表3。由表3可知,其他条件一定时,当随着BOP的投料量增加至1.2时,产物收率也慢慢提高。因此,n(6) ∶n(BOP)为1 ∶1.2时,比较适宜,收率较高(57.6%)。

表3 BOP用量对加成反应的影响
(4) 7的合成
在7的合成中,考察了反应时间的影响,结果见表4。由表4可知,当反应时间增加至10 h时收率最高(87%),之后逐渐趋于平稳,且不再增加,因此反应10 h为宜。

表4 反应时间对环合反应的影响
(5) 8的合成
在8的合成中,考擦了NaOH和(BOC)2O的投料量对加成反应的影响,结果见表5。由表5可知,n(7)∶n(NaOH) ∶n[(BOC)2O]为1 ∶2 ∶2.2时,收率最高(92.6%)。

表5 NaOH和(BOC)2O的用量对反应的影响
以丙二酸二乙酯为原料,经8步反应合成了4,7-二氮杂螺[2.5]辛烷-7-甲酸,总收率9.16%。该合成路线避免使用常温下性质不稳定、价格昂贵、安全性差的原料,避免了苛刻的设备要求条件,具有成本低、操作简单等优点,具有较大的工业化应用前景。
[1] 方巍. 中国医药、农药和染料中间体市场现状及发展趋势[J].精细化工原料及中间体,2008,09:7-10.
[2] 徐兆瑜. 医药中间体的发展和市场前景(二)[J]. 化工中间体,2003,13:1-5,9.
[3] 张晓琴,章杰. 亚洲有机中间体发展和市场现状分析[J].精细化工中间体,2002,01:3-7,49.
[4] 高洪建,余中心. 抗菌药物中间体开发研究[J].精细化工中间体,2001,31(2):1-6.
[5] 王荣耕. 与β-内酰胺抗生素相关的精细化工中间体[J].精细与专用化学品,2000,8(13):9-11.
[6] 王明学. 我国医药产销形势和重要中间体[J]. 精细与专用化学品,2001,12:3-6.
[7] 章炜,徐亮. 新型2,4-二取代四氢吡咯-3-羧酸衍生物的合成[J].合成化学,2016,24(1):43-46.
[8] 张丽慧,姜帆,徐晓剑,等. 新型1,3,5-三嗪衍生物的合成[J].合成化学,2014,22(6):507-509.
[9] 陈志卫,陈君培. (S,S)-2,8-二氮杂双环[4,3,0]壬烷的合成工艺改进[J].合成化学,2013,21(6):760-762.
[10] 敖聪聪. 世界药物研究发展趋势与市场预测[J].精细化工中间体,2004,34:5-8.
[11] 周文华. 杨辉荣绿化学技术在制药工业中的应用[J].上海化工,2004,20:29.
[12] Terada T, Noda S, Inui K I. Management of dose variability and side effects for individualized cancer pharmacotherapy with tyrosine kinase inhibitors[J]. Pharmacology & Therapeutics,2015,152:125-134.
[13] Dean D K, Takle A K, Wilson D M. Imidazole derivatives as Raf kinase inhibitors: CA, EP 1 318 992 A1[P].2003.
[14] 李晓红,邓如伟,余蓉,等. 猪胰酶、胰岛素与胰蛋白酶抑制剂的联产[J].华西药学杂志,2009,24(5):480-482.
[15] Chang X W, Han Q C, Jiao Z G,etal. 1-Aminoxymethylcyclopropanecarboxylic acid as building block ofβN—O turn and helix:Synthesis and conformational analysis in solution and in the solid state[J].Tetrahedron,2010,66(51):9733-9737.
[16] Chang X W, Han Q C, Jiao Z G,etal. 1-Aminoxymethylcyclopropanecarboxylic acid as building block ofβN—O turn and helix:Synthesis and conformational analysis in solution and in the solid state[J].Tetrahedron,2010,66(51):9733-9737.[17] Kiely J S, Schroeder M C, Sesnie J C. New “ofloxacin” type antibacterial agents. Incorporation of the spiro cyclopropyl group atN-1[J].Journal of Medicinal Chemistry,1988,31(10):2004-2008.
[18] Wheeler T N, Ray J A. A convenient and efficient synthesis of 1-aminocyclopropanecarboxylic acid (ACC)[J].Cheminform,1988,19(36):141-149.
[19] Abou-Donia M M, Wilson S P , Zimmerman T P,etal. Regulation of guanosine triphosphate cyclohydrolase and tetrahydrobiopterin levels and the role of the cofactor in tyrosine hydroxylation in primary cultures of adrenomedullary chromaffin cells[J].Journal of Neurochemistry,1986,46(4):1190-1199.
[20] Brackmann F, Es-Sayed M, De Meijere A. Synthesis of spirocyclopropanated analogues of iprodione[J].Annalen Der Chemie Und Pharmacie,2005,2005(2005):2250-2258.
[21] Ohya S, Matsuo M. Colored polyimide molded article,and process for production thereof:WO 2 011 043 467 A1[P].2011.
[22] Chu D T W, Claiborne A K, Clement J J,etal. Syntheses and antibacterial activity of novel 6-fluoro-7-(gem-disubstituted piperazin-1-yl)-quinolines[J].Canadian Journal of Chemistry,2011,70(5):1328-1337.
Synthesis 4,7-Two Aza Spiro[2.5]Octane-7-formic Acid Tert Butyl Ester
FENG Jin-jin1, ZHU Hui2, WANG Hui-dong1,CHEN Chu-jun1, LIU Bao-gui1, HUANG Qi-mao1*
(1. College of Chemical and Pharmaceutical, Wuhan Institute of Technology, Wuhan 430073, China;2. Wuhan Puren Hospital, Wuhan 430073, China)
4,7-Two[2.5] octane-7-tert butyl ester with the total yiled of 9.16% was syhthesized by cyclization reaction, Hoffman reaction, hydrolysis, acylation, etc, using the diethyl malonate as raw material. The structure was confirmed by1H NMR and MS.
diethylmalonate; 4,7-twoazaspiro[2.5]octane-7-formicacidtertbutylester; Hoffman reaction; drug synthesis
2016-04-28;
2016-09-05
冯瑾瑾(1988-),女,汉族,湖北枣阳人,硕士研究生,主要从事药物中间体的合成研究。 E-mail: 2413630422@qq.com
黄齐茂,博士生导师, E-mail: huangqim@163.com
R914.5; O621.3
A
10.15952/j.cnki.cjsc.1005-1511.2016.10.16113
