不同纤维来源饲粮和细胞壁降解酶对猪肠道微生物菌群多样性的影响
2016-11-15张永婧张文明张志静王伟兰
张永婧 刘 强 张文明 张志静 王伟兰 庄 苏
(南京农业大学动物科技学院,南京210095)
不同纤维来源饲粮和细胞壁降解酶对猪肠道微生物菌群多样性的影响
张永婧刘强*张文明张志静王伟兰庄苏
(南京农业大学动物科技学院,南京210095)
本试验旨在利用末端限制性片段长度多态性技术研究不同纤维来源饲粮和细胞壁降解酶对猪肠道微生物菌群多样性及其组成结构的影响。试验选用8头平均体重为(35.0±2.5) kg的“杜×长×大”三元杂交生长猪,统一安装回肠末端T型瘘管,随机分为4组,每组2个重复。试验分4期,每期每组猪按4×4拉丁方设计饲喂小麦麸、小麦麸加酶、大豆皮和大豆皮加酶4种试验饲粮之一。每期预试期15 d,正试期6 d。试验结果表明:1)大豆皮饲粮组猪回肠食糜微生物的多样性显著高于小麦麸饲粮组(P<0.05)。2)小麦麸饲粮组显著提高猪回肠食糜中普氏菌属(Prevotella)和乳酸杆菌属(Lactobacillus)的丰度(P<0.05),而大豆皮饲粮组则显著提高猪回肠食糜中瘤胃球菌属(Ruminococcus)及粪便中拟杆菌属(Bacteroides)、毛螺菌属(Lachnospira)的丰度(P<0.05)。3)添加细胞壁降解酶显著提高各饲粮组猪回肠食糜中Lachnospira、真细菌属(Eubacterium)及粪便中Lachnospira的丰度(P<0.05),但也同时降低了回肠食糜中Lactobacillus、Prevotella及粪便中Lactobacillus、Bacteroides和克雷伯氏菌属(Klebsiella)的丰度(P<0.05)。综上所述,纤维饲粮可显著提高猪肠道内非淀粉多糖降解菌的丰度,而细胞壁降解酶则可选择性改变肠道微生物菌群的多样性及其组成。
生长猪;饲粮纤维;细胞壁降解酶;肠道微生物;末端限制性片段长度多态性
猪肠道内寄居着种群庞杂的微生物[1],肠道微生物除了发酵在小肠内未消化吸收的碳水化合物和蛋白质等养分,产生短链脂肪酸为猪提供能量之外,还通过维生素和酶合成、胆汁酸盐重吸收、促进消化道发育及其免疫系统成熟等多种方式影响猪的营养与健康[2-3]。研究表明,饲粮对猪肠道微生物菌群结构有明显影响,其中饲粮纤维的数量和类型备受关注[4]。谷物及其副产物中细胞壁非淀粉多糖(non-starch polysaccharides,NSP)是猪饲粮的主要纤维来源,其含量、组成和化学结构因植物种类、组织和生长阶段而异[5-6]。现有同类研究多基于多种纤维来源的混合饲粮[2,4,7],而对单一纤维来源饲粮的研究较少,不利于对饲粮NSP化学特性与猪消化道微生物菌群结构间构效关系的认识。
饲粮纤维通常不能被猪的内源消化酶降解,而且还会影响其他养分的消化利用,被视为一类抗营养因子[8]。养猪和饲料生产实践中,消除饲粮纤维副作用最常见的对策是添加NSP降解酶等外源酶制剂[9-10]。Bedford等[11]研究表明,外源NSP降解酶可促进不溶性NSP降解,生成可发酵寡糖,进而影响肠道微生物组成。然而,产生的寡糖结构又随饲粮NSP组成和外加NSP降解酶种类而异。同时,肠道微生物对寡糖的利用能力也存在着明显的种属差异性[12-14]。因此,本试验旨在利用末端限制性片段长度多态性(terminal restriction fragment length polymorphism,T-RFLP)技术研究纤维来源(小麦麸或大豆皮)以及添加外源细胞壁降解酶对猪肠道微生物菌群多样性的影响,为深入了解饲粮纤维组成与猪肠道微生物菌群结构间的构效关系,以及细胞壁降解酶在谷物副产物饲用开发中的应用潜力提供参考。
1 材料与方法
1.1试验设计
试验选用8头平均体重为(35.0±2.5) kg的“杜×长×大”三元杂交生长猪,统一安装回肠末端T型瘘管(回盲瓣前15~20 cm),随机分为4组,每组2个重复。试验分4期,每期每组试验猪按4×4拉丁方设计安排饲喂1种试验饲粮,每个试验期21 d,其中前15 d为预试期,饲喂全价配合饲粮(购自江苏海普瑞饲料有限公司),预试期第1天试验饲粮替换部分全价配合饲粮,5 d内全部替换为试验饲粮,其余10 d用于猪适应试验饲粮并维持肠道菌群的稳定。第16~18天肛门采集粪便样品,第19~21天11:00和14:00采集回肠食糜样品。样品用无菌袋收集,封装在经高温灭菌的5 mL塑料离心管内,-80 ℃冻存。T-RFLP分析前,将样品室温消融,分别等量混合每个试验期3 d采集的饲喂同一种饲粮的2头猪的回肠食糜和粪便样品,作为分析试样。
1.2饲粮组成及营养水平
试验饲粮参照我国《瘦肉型猪饲养标准》(2004年版)配制,分别以小麦麸和大豆皮[均购自东海粮油(张家港)有限公司]作为单一纤维源,辅以玉米淀粉、鱼粉、血粉和指示剂三氧化二铬(Cr2O3)等配成2种营养均衡,能量、蛋白质、纤维水平相近的半纯合基础饲粮,分别为小麦麸饲粮(wheat bran diet,WB)和大豆皮饲粮(soybean hull diet,SH),其组成及营养水平见表1。在2种基础饲粮中分别添加相同剂量的混合酶制剂(纤维素酶1 000 U/kg、木聚糖酶5 000 U/kg和植酸酶1 500 U/kg),共形成4种试验饲粮。
1.3检测指标
1.3.1DNA提取
参考Bindelle等[15]的方法提取肠道微生物总DNA。

表1 基础饲粮组成及营养水平(风干基础)
1)维生素预混料为每千克饲粮提供 The vitamin premix provided the following per kg of diets:VA 10 000 IU,VD34 000 IU,VE 32 mg,VK34 mg,VB13 mg,VB28 mg,VB65 mg,VB120.05 mg,烟酸 nicotinic acid 45 mg,D-泛酸D-pantothenic acid 20 mg,叶酸 folic acid 2 mg,生物素 biotin 0.15 mg,氯化胆碱 choline chloride 360 mg。
2)微量元素预混料为每千克饲粮提供 The mineral premix provided the following per kg of diets:Fe 150 mg,Cu 8 mg,Zn 100 mg,Mn 50 mg,I 0.3 mg,Se 0.3 mg。
3)消化能为计算值,其余为实测值。DE was a calculated value and the others were measured values.
1.3.2PCR扩增
试验选用16S rRNA全序列引物:8F:5’-AGAGTTTGATCCTGGCTCAG-3’和1492R:5’-GGTTACCTTGTTACGACTT-3’。PCR(ABI VeritiTM96-well Thermal Cycler,美国ABI公司)反应体系共25 μL,含2种引物各0.5 μL,DNA模板1.0 μL,Taq DNA聚合酶12.5 μL,无菌水10.5 μL。反应程序为:94 ℃预变性5 min,94 ℃变性20 s,55 ℃退火30 s,72 ℃延伸50 s,共35个循环,最后72 ℃延伸7 min。取5.0 μL扩增产物进行1%琼脂糖凝胶电泳(EPS604电泳仪,南京科室仪器研究所研制)检测,预期扩增产物大小约1 500 bp。
1.3.3限制性内切酶酶切
利用AxyPrep PCR产物纯化试剂盒(美国Axygen公司)对PCR产物进行纯化,而后各取5 μL纯化产物,采用限制性内切酶HhaI(日本Takara公司1056A)进行酶切。酶切体系为20 μL:PCR纯化产物5 μL,10 mol/L Buffer 2 μL,内切酶 1 μL,灭菌水12 μL。37 ℃酶切2 h,65 ℃灭活30 min。
1.3.4酶切产物毛细管电泳
纯化后的酶切产物在DNA测序仪(美国ABI公司ABI 3730XL DNA Analyzer)上进行毛细管电泳(样品送上海生工生物工程技术服务有限公司检测)。
1.4数据处理与分析
电泳条带利用GeneMarker V2.2.0软件进行定性和定量分析,获得样品各个末端限制性片段(terminal restriction fragments,T-RFs)的长度、峰高及峰面积等参数。剔除长度小于50 bp和大于600 bp的片段,并根据片段峰高计算微生物菌群的多样性指数。利用MICA: T-RFLP Analysis[16]软件在线分析片段所对应的微生物种属。采用SPSS 18.0中GLM模型法对多样性指数和各片段相对含量进行2因素完全因子ANOVA分析,比较不同纤维饲粮组间微生物组成的差异。P<0.05时表示差异显著,P<0.10时表示有差异趋势。
微生物菌群多样性指数计算公式及其定义如下:
物种丰度(表示微生物菌群中
物种的个数)=限制性片段的个数;
香侬威纳指数(表示微生物菌群组成的复杂程度,
该指数越大,表明菌群的组成结构越复杂)=
-∑PilnPi;
辛普森指数(表示菌群中优势菌属
所占比重及菌群的均衡度,该指数越大,
表明优势菌属所占的比例越大,菌群均衡度则
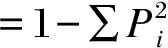
式中:Pi表示第i个峰的峰高占总峰高的比例。
2 结果与分析
2.1微生物菌群多样性
经软件分析,本试验平均每头猪回肠末端食糜和粪便样品中获得长度在50~600 bp的酶切片段分别为21和35个,其中相对含量在1%以上的片段分别为13和17个。各饲粮组猪回肠末端食糜和粪便中微生物菌群的多样性指数见表2。SH组猪回肠末端的微生物丰度显著高于WB组(P<0.05),但纤维来源对猪粪便微生物的物种丰度及香侬威纳指数均无显著影响(P>0.05)。添加酶制剂有提高SH组猪回肠食糜微生物菌群香侬威纳指数的趋势(P<0.10)。各饲粮组猪回肠食糜微生物菌群的辛普森指数受纤维来源与酶制剂之间交互作用的影响(P<0.05)。
2.2猪回肠末端食糜微生物的丰度
试验获得相对含量大于1%的回肠末端食糜微生物T-RFs共13个,其所代表菌属及相对含量见表3。WB组显著提高猪回肠末端食糜微生物菌群中乳酸杆菌属(Lactobacillus)和普氏菌属(Prevotella)的丰度(P<0.05),而SH组则显著提高猪回肠中瘤胃球菌属(Ruminococcus)的丰度(P<0.05)。添加酶制剂显著提高各饲粮组猪回肠中变形菌门(Proteobacteria)的未知菌、毛螺菌属(Lachnospira)和真细菌属(Eubacterium)的丰度(P<0.05),但显著降低WB组猪回肠中Lactobacillus和Prevotella的丰度(P<0.05)。各饲粮组猪回肠食糜微生物菌群中Lactobacillus和Prevotella的丰度受纤维来源与酶制剂之间交互作用的影响(P<0.05)。
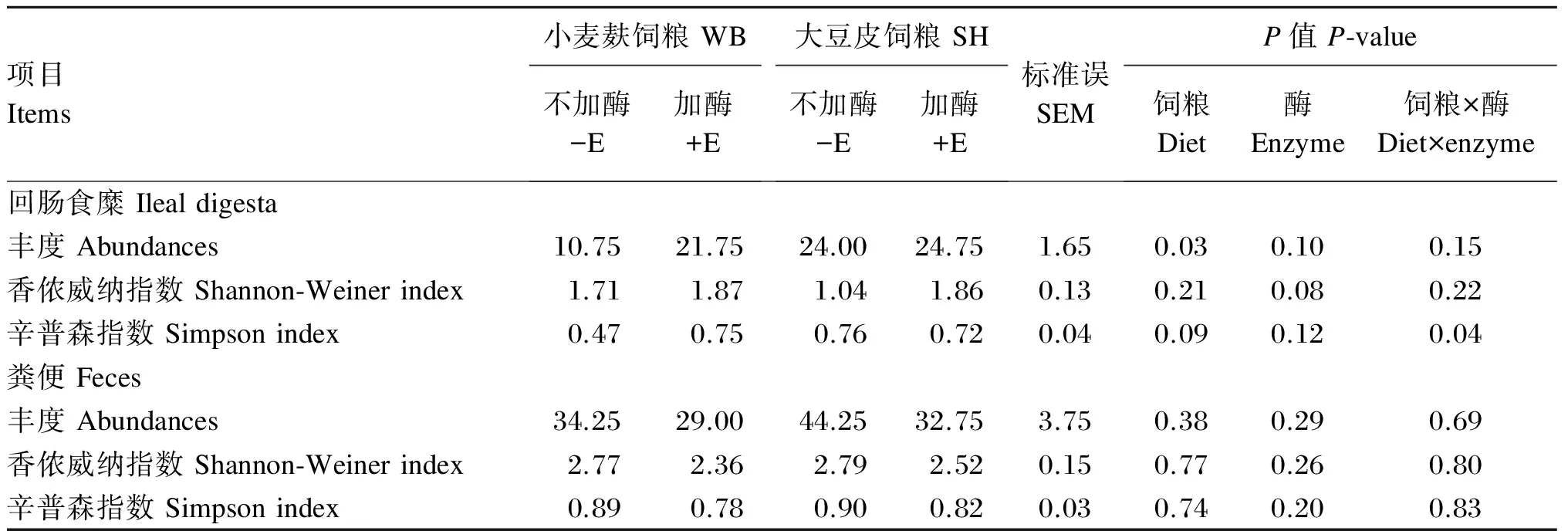
表2 各饲粮组猪回肠末端食糜和粪便样品中微生物菌群的多样性指数
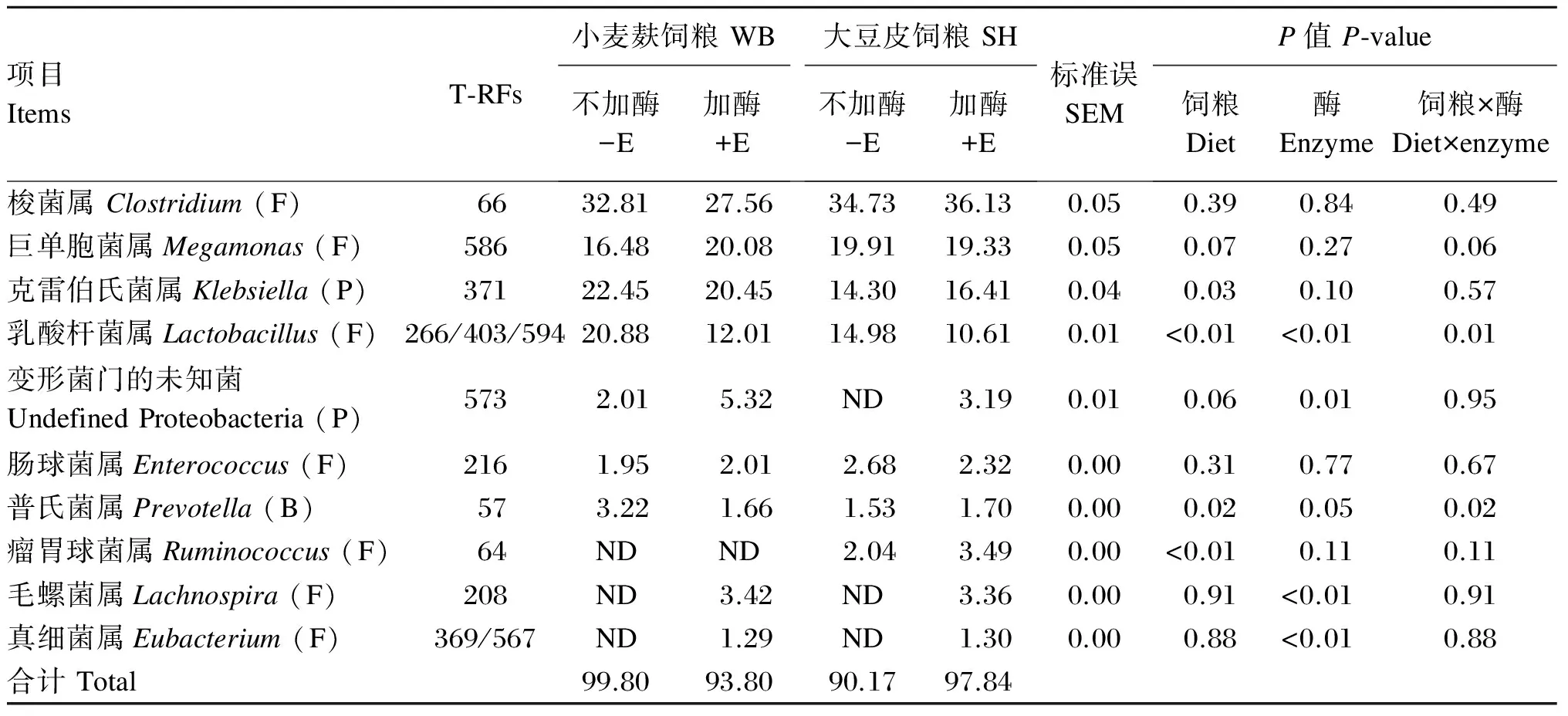
表3 各饲粮组猪回肠末端食糜微生物的丰度
T-RFs:末端限制性片段;ND:未检测出;F:厚壁菌门;P:变形菌门;B:拟杆菌门。下表同。
T-RFs: terminal restriction fragments; ND: not detected; F: Firmicutes; P: Proteobacteria; B: Bacteroidetes. The same as below.
2.3猪粪便微生物的丰度
试验获得相对含量大于1%的粪便微生物T-RFs共17个,其所代表菌属及相对含量见表4。WB组显著提高猪粪便微生物菌群中巨单胞菌属(Megamonas)和光岗菌属(Mitsuokella)的丰度(P<0.05),Lactobacillus和克雷伯氏菌属(Klebsiella)的丰度相比于SH组有升高趋势(P<0.10)。SH组则显著提高猪粪便中拟杆菌属(Bacteroides)和Lachnospira的丰度(P<0.05)。添加酶制剂显著提高各饲粮组猪粪便中Proteobacteria内未知菌和Lachnospira的丰度(P<0.05),同时显著降低Lactobacillus和Klebsiella的丰度(P<0.05),以及SH组猪粪便中Bacteroides的丰度(P<0.05)。各饲粮组猪粪便微生物菌群中Eubacterium的丰度受纤维来源与酶制剂之间交互作用的影响(P<0.05)。
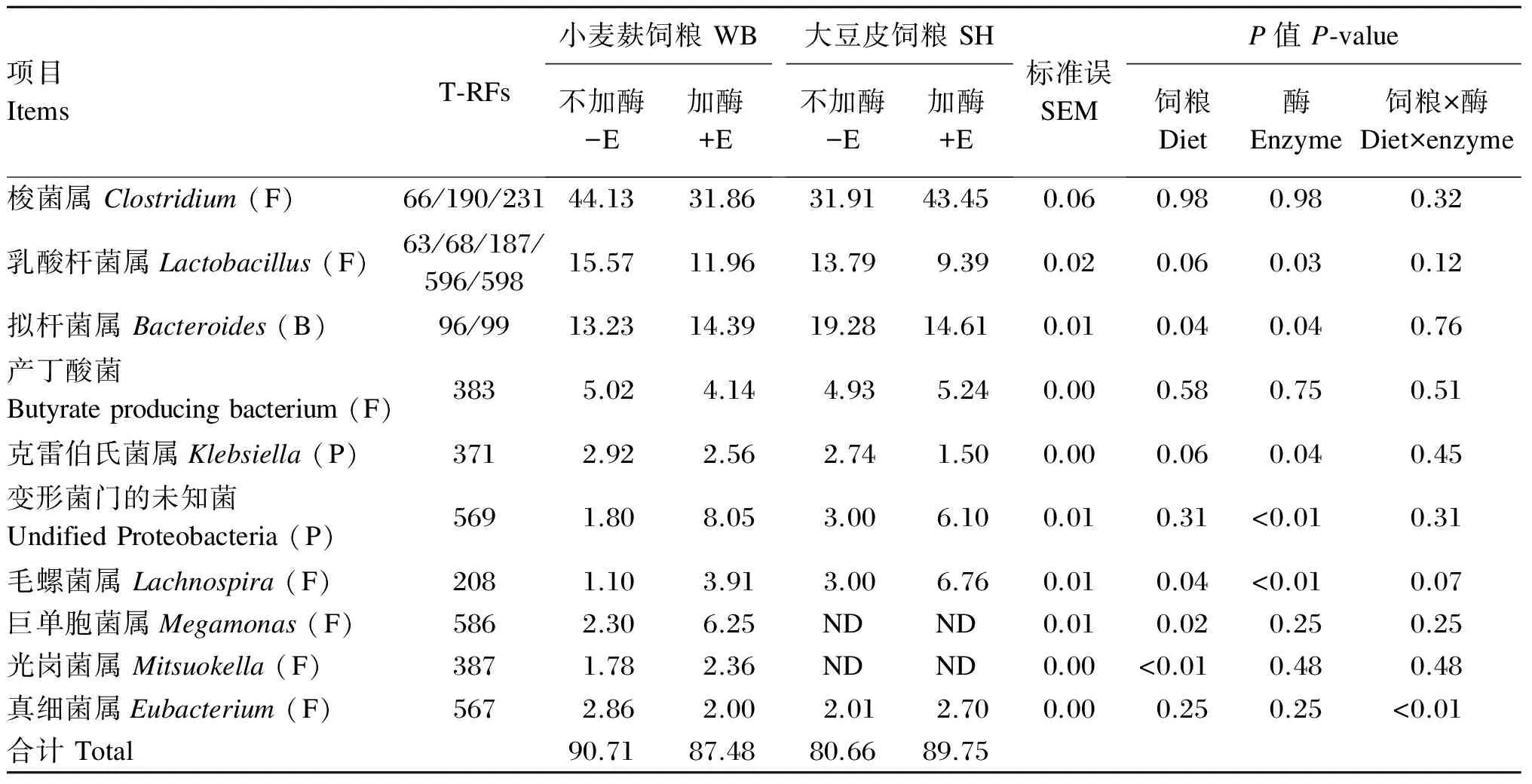
表4 各饲粮组猪粪便微生物的丰度
3 讨 论
3.1不同纤维来源饲粮和细胞壁降解酶对猪肠道微生物菌群多样性的影响
总体而言,本试验猪粪便样品微生物的物种丰度明显高于回肠末端食糜样品,物种丰度越高表明该菌群的多样性越高,其组成结构越复杂,相应地该菌群中优势菌属的比重则越低。饲粮纤维来源和酶制剂对猪回肠食糜微生物多样性有不同程度的影响,但对粪便微生物的多样性相关指数均无显著影响。该结果表明猪小肠微生物区系更易受饲粮因素的影响,而大肠微生物区系因其组成结构复杂且均衡度高则相对稳定。一般来讲,消化道微生物的生长发育与饲粮中可利用底物的种类和数量有一定关系[17]。本试验2种纤维饲粮的中性洗涤纤维含量虽然接近,但WB的NSP主要由阿拉伯糖、木糖和葡萄糖(阿拉伯木聚糖和纤维素)组成,而SH除此之外还含有较高的糖醛酸(果胶类物质),总NSP含量也高于WB。此外,大豆皮中可溶性NSP的含量也显著高于小麦麸[2,6]。虽然Liu等[4]表明高果胶饲粮和高阿拉伯木聚糖饲粮并未对猪肠道微生物多样性产生显著影响,但Ivarsson[1]却发现小麦麸饲粮会刺激猪回肠微生物菌群中Lactobacillus的生长,且推测该菌能产生抗菌化合物,抑制其他菌种繁衍,从而导致猪肠道微生物多样性的下降。本试验WB组猪回肠末端食糜微生物菌群的丰度显著低于SH组,也可能与WB可利用底物的多样性及阿拉伯木聚糖选择性刺激Lactobacillus的生长有关。
3.2不同纤维来源饲粮和细胞壁降解酶对猪肠道微生物菌群组成结构的影响
Lamendella等[18]指出,猪肠道内的微生物多与碳水化合物代谢有关,其中主要为革兰氏阳性菌厚壁菌门(Firmicutes)和革兰氏阴性菌拟杆菌门(Bacteroidetes)。本试验鉴定为梭菌属(Clostridium)、Lactobacillus、肠球菌属(Enterococcus)、Eubacterium、Megamonas、Mitsuokella、Lachnospira和Ruminococcus等菌属的T-RFs均属于Firmicutes,总平均丰度分别占回肠食糜和粪便微生物总量的72%和65%,该结果与Lamendella等[18]和Niu等[19]在猪肠道内的研究结果相似。研究表明,大多数Firmicutes细菌能分泌胞外多糖降解酶,部分菌属(如Ruminococcus和Clostridium)具有结构复杂的多功能纤维小体,既能降解不溶性的木葡聚糖和木聚糖等半纤维素,也能有效降解纤维素,释放可溶性寡糖,但个别菌种[如热纤梭菌(R.thermocellum)]并不能利用其戊聚糖降解产物[20-21]。而Bacteroidetes细菌(如Prevotella)的糖降解代谢系统复杂且全面,易受底物调控,适应能力极强,主要参与降解寡糖、淀粉、果胶、木葡聚糖和木聚糖等多种可溶性多糖,但对纤维素降解能力较差[22]。本试验所得猪肠道细菌绝大多数来自Firmicutes,而Bacteroidetes细菌丰度相对较低(1.53%~19.28%),可能与WB和SH中NSP均以不溶性多糖为主(分别占总NSP的84%和77%)有关。然而从小肠到大肠,Bacteroidetes细菌的丰度明显升高,这可能是由于在细菌和外源多糖酶制剂的作用下,饲粮NSP的溶解量增加或生成可溶性寡糖[23],为Bacteroidetes细菌提供适宜的可利用底物。本试验所有组猪回肠末端食糜中检测出的Proteobacteria细菌丰度都明显高于同类研究。例如,在前人的研究中Klebsiella与肠道木聚糖的降解有关,但相对丰度较低[24]。Proteobacteria细菌多数与蛋白质发酵有关[25],该菌门丰度升高的原因可能与本试验为平衡各纤维饲粮的蛋白质与能量比,添加了较高含量的鱼粉和血浆蛋白粉等动物蛋白质原料有关。随着绝大多数饲粮蛋白质在小肠中被消化[26],该门细菌的丰度从回肠食糜到粪便显著下降,这也间接证明了上述推测。同理,随着饲粮淀粉在回肠之前几乎被完全消化[26],各试验组猪粪便中Lactobacillus和Enterococcus(肠道内主要淀粉降解菌[2,27])的丰度也较回肠食糜明显下降,粪便中甚至均未检测出Enterococcus。可见,可利用养分对肠道菌群组成可能起着决定性作用。
肠道微生物对NSP的降解程度既取决于其自身的理化特性(如单糖组成、聚合度和溶解性等)[28],也与肠道微生物菌属间的互作有关[1]。研究表明,Clostridium菌属是人类肠道中主要的纤维降解菌,与Eubacterium和Prevotella等菌属互作可有效降解纤维素及半纤维素[29-31]。本试验各饲粮组猪肠道中均以Clostridium相对丰度最高,且不受纤维来源和外源酶制剂的影响。Lactobacillus和Bacteroides的丰度次之,但前者在WB组猪回肠食糜和粪便中的丰度均显著高于SH组,而后者仅在粪便中出现,且SH组中的丰度高于WB组。Sanchez等[32]研究表明,某些Lactobacillus菌种能分泌阿拉伯木聚糖水解酶,在木寡糖或阿拉伯木聚糖培养基内生长。Ivarsson[1]也报道,麦麸可促进猪肠道内Lactobacillus的生长,与本试验的结果基本吻合。同为Bacteroidetes的Prevotella和Bacteroides,前者仅出现在回肠食糜中,而后者仅出现在粪便中,可能与Bacteroides主要利用可溶性多糖和寡糖有关[28]。同为乳酸利用菌的Megamonas和Mitsuokella,前者在回肠食糜中的丰度相当高,而在粪便中显著下降;而Mitsuokella仅在粪便中出现,同时伴随着一种未知丁酸生成菌(产丁酸菌)的出现,可能是肠道微生物之间在底物降解和产物利用方面存在互作关系的体现。据Weber等[33]报道,Lachnospira为人类肠道的主要果胶降解菌。该属细菌在本试验SH组猪粪便中的丰度显著高于WB组,与SH的果胶含量高于WB相对应。未添加外源酶制剂时,表3中各饲粮组猪回肠食糜中均未检测出Lachnospira,而添加酶制剂后其丰度均显著升高,说明添加细胞壁降解酶促进原料中果胶的溶解或降解。但是无论WB或SH,其中的果胶成分主要在猪后肠发酵[34-35],因此粪便中该菌属的丰度显著高于回肠食糜。类似地,由于外源细胞壁降解酶加快淀粉在猪回肠之前的消化[11],减少了淀粉降解菌Lactobacillus的可利用底物,使其丰度下降,表现出Bedford等[11]所谓的外源酶制剂对小肠内淀粉降解菌生长的抑制现象。
尽管T-RFLP技术具有快速、低成本和高通量的优点,但仅依据限制性酶切片段长度而非其碱基序列信息不足以准确区分微生物的种类。根据其原理可知,该方法所得结果可能受内切酶的种类和作用时间、毛细管电泳图谱质量以及数据库范围等因素的影响,导致菌属间的归属出现误差。例如,片段T-RF 370在Jensen等[36]的研究中被归属于肠杆菌属(Enterobacter),而与之仅有1个碱基长度差的T-RF 371在本试验中则被归属于Klebsiella。为了验证T-RFLP结果的可靠性,本次试验进一步对样品进行了Illumina MiSeq高通量测序。初步统计结果表明,虽然高通量测序技术可鉴定出更多的微生物种类,且能鉴定到属水平,但二者所得微生物菌群的多样性和与纤维相关的优势菌属基本一致。本文基于T-RFLP技术所得结果,在反映肠道微生物菌群多样性方面也具有一定的参考价值。
4 结 论
① T-RFLP检测结果表明,纤维来源显著影响猪回肠末端食糜微生物菌群的多样性,但对粪便微生物的多样性无显著影响。
② 小麦麸饲粮可显著提高猪肠道微生物菌群中半纤维素降解菌Lactobacillus、Prevotella、Megamonas和Mitsuokella等的丰度,而大豆皮饲粮则显著提高猪肠道中果胶降解菌Lachnospira、纤维素降解菌Ruminococcus和Bacteroides等的丰度。
③ 添加酶制剂选择性地影响猪肠道微生物菌群中Lachnospira、Eubacterium、Lactobacillus、Prevotella和Bacteroides等的丰度。
[1]IVARSSON E.Chicory (CichoriumintybusL.) as fibre source in pig diets-effects on digestibility,gut microbiota and performance[D].Ph.D.Thesis.Uppsala:Swedish University of Agricultural Sciences,2012.
[2]IVARSSON E,ROOS S,LIU H Y,et al.Fermentable non-starch polysaccharides increases the abundance ofBacteroides-Prevotella-Porphyromonasin ileal microbial community of growing pigs[J].Animal,2014,8(11):1777-1787.
[3]CUMMINGS J H.Short chain fatty acids in the human colon[J].Gut,1981,22(9):763-779.
[4]LIU H Y,IVARSSON E,LUNDH T,et al.Chicory (CichoriumintybusL.) and cereals differently affect gut development in broiler chickens and young pigs[J].Journal of Animal Science and Biotechnology,2013,4:50.
[5]KNUDSEN K E B.Carbohydrate and lignin contents of plant materials used in animal feeding[J].Animal Feed Science and Technology,1997,67(4):319-338.
[6]LATTIMER J M,HAUB M D.Effects of dietary fiber and its components on metabolic health[J].Nutrients,2010,2(12):1266-1289.
[7]MONTAGNE L,PLUSKE J R,HAMPSON D J.A review of interactions between dietary fibre and the intestinal mucosa,and their consequences on digestive health in young non-ruminant animals[J].Animal Feed Science and Technology,2003,108(1/2/3/4):95-117.
[8]WILFART A,MONTAGNE L,SIMMINS P H,et al.Sites of nutrient digestion in growing pigs:effect of dietary fiber[J].Journal of Animal Science,2007,85(4):976-983.
[9]JIA W,SLOMINSKI B A,BRUCE H L,et al.Effects of diet type and enzyme addition on growth performance and gut health of broiler chickens during subclinicalClostridiumperfringenschallenge[J].Poultry Science,2009,88(1):132-140.
[10]L☞RKE H N,ARENT S,DALSGAARD S,et al.Effect of xylanases on ileal viscosity,intestinal fiber modification,and apparent ileal fiber and nutrient digestibility of rye and wheat in growing pigs[J].Journal of Animal Science,2015,93(9):4323-4335.
[11]BEDFORD M R,COWIESON A J.Exogenous enzymes and their effects on intestinal microbiology[J].Animal Feed Science and Technology,2012,173(1/2):76-85.
[12]FLINT H J,SCOTT K P,DUNCAN S H,et al.Microbial degradation of complex carbohydrates in the gut[J].Gut Microbes,2012,3(4):289-306.
[13]HAMAKER B R,TUNCIL Y E.A perspective on the complexity of dietary fiber structures and their potential effect on the gut microbiota[J].Journal of Molecular Biology,2014,426(23):3838-3850.
[14]VAN DER MEULEN J,INBORR J,BARKER J G M.Effects of cell wall degrading enzymes on carbohydrate fractions and metabolites in stomach and ileum of pigs fed wheat bran based diets[J].Archiv für Tierernaehrung,2001,54(2):101-115.
[15]BINDELLE J,PIEPER R,MONTOYA C A,et al.Nonstarch polysaccharide-degrading enzymes alter the microbial community and the fermentation patterns of barley cultivars and wheat products in aninvitromodel of the porcine gastrointestinal tract[J].FEMS Microbiology Ecology,2011,76(3):553-563.
[16]SHYU C,SOULE T,BENT S J,et al.MiCA:a web-based tool for the analysis of microbial communities based on terminal-restriction fragment length polymorphisms of 16S and 18S rRNA genes[J].Microbial Ecology,2007,53(4):562-570.
[17]ROCA-CANUDAS M,ANGUITA M,NOFRARAS M,et al.Effects of different types of dietary non-digestible carbohydrates on the physico-chemical properties and microbiota of proximal colon digesta of growing pigs[J].Livestock Science,2007,109(1/2/3):85-88.
[18]LAMENDELLA R,SANTO DOMINGO J W,GHOSH S,et al.Comparative fecal metagenomics unveils unique functional capacity of the swine gut[J].BMC Microbiology,2011,11:103.
[19]NIU Q,LI P H,HAO S S,et al.Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs[J].Scientific Reports,2015,5:9938.
[20]WHITE B A,LAMED R,BAYER E A,et al.Biomass utilization by gut microbiomes[J].Annual Review of Microbiology,2014,68(1):279-296.
[21]RAVACHOL J,DE PHILIP P,BORNE R,et al.Mechanisms involved in xyloglucan catabolism by the cellulosome-producing bacteriumRuminiclostridiumcellulolyticum[J].Scientific Reports,2016,6:22770.
[22]LARSBRINK J,ROGERS T E,HEMSWORTH G R,et al.A discrete genetic locus confers xyloglucan metabolism in select human gutBacteroidetes[J].Nature,2014,506(7489):498-502.
[23]ZENED A,COMBES S,CAUQUIL L,et al.Microbial ecology of the rumen evaluated by 454 GS FLX pyrosequencing is affected by starch and oil supplementation of diets[J].FEMS Microbiology Ecology,2013,83(2):504-514.
[24]DODD D,MACKIE R I,CANN I K O.Xylan degradation,a metabolic property shared by rumen and human colonicBacteroidetes[J].Molecular Microbiology,2011,79(2):292-304.
[25]COWIESON A J,BEDFORD M R.The effect of phytase and carbohydrase on ileal amino acid digestibility in monogastric diets:complimentary mode of action?[J].World’s Poultry Science Journal,2009,65(4):609-624.
[26]高月琴,王伟兰,张亚伟,等.不同来源纤维在生长猪消化道内的食糜理化特性和养分消化率[J].南京农业大学学报,2015,38(3):471-477.
[27]REGMI P R,METZLER-ZEBELI B U,GNZLE M G,et al.Starch with high amylose content and lowinvitrodigestibility increases intestinal nutrient flow and microbial fermentation and selectively promotesBifidobacteriain pigs[J].The Journal of Nutrition,2011,141(7):1273-1280.
[28]FLINT H J,BAYER E A,RINCON M T,et al.Polysaccharide utilization by gut bacteria:potential for new insights from genomic analysis[J].Nature Reviews Microbiology,2008,6(2):121-131.
[29]WEDEKIND K J,MANSFIELD H R,MONTGOMERY L.Enumeration and isolation of cellulolytic and hemicellulolytic bacteria from human feces[J].Applied and Environmental Microbiology,1988,54(6):1530-1535.
[30]YODA K,TOYODA A,MUKOYAMA Y,et al.Cloning,sequencing,and expression of aEubacteriumcellulosolvens 5 gene encoding an endoglucanase (Cel5A) with novel carbohydrate-binding modules,and properties of Cel5A[J].Applied and Environmental Microbiology,2005,71(10):5787-5793.
[31]NIELSEN T S,L☞RKE H N,THEIL P K,et al.Diets high in resistant starch and arabinoxylan modulate digestion processes and SCFA pool size in the large intestine and faecal microbial composition in pigs[J].British Journal of Nutrition,2014,112(11):1837-1849.
[32]SANCHEZ J I,MARZORATI M,GROOTAERT C,et al.Arabinoxylan-oligosaccharides (AXOS) affect the protein/carbohydrate fermentation balance and microbial population dynamics of the simulator of human intestinal microbial ecosystem[J].Microbial Biotechnology,2009,2(1):101-113.
[33]WEBER F H,CANALE-PAROLA E.Pectinolytic enzymes of oral spirochetes from humans[J].Applied and Environmental Microbiology,1984,48(1):61-67.
[34]IVARSSON E,ANDERSSON R,LINDBERG J E.Digestibility of fibre sources and molecular weight distribution of fibre fractions in ileal digesta of growing pigs[J].Archives of Animal Nutrition,2012,66(6):445-457.
[35]JHA R,LETERME P.Feed ingredients differing in fermentable fibre and indigestible protein content affect fermentation metabolites and faecal nitrogen excretion in growing pigs[J].Animal,2012,6(4):603-611.
[36]JENSEN A N,MEJER H,MØLBAK L,et al.The effect of a diet with fructan-rich chicory roots on intestinal helminths and microbiota with special focus onBifidobacteriaandCampylobacterin piglets around weaning[J].Animal,2011,5(6):851-860.
(责任编辑武海龙)
Gastrointestinal Microbial Flora Diversity in Pigs Fed Different Fibrous Diets with or without Cell Wall-Degrading Enzyme Supplementation
ZHANG YongjingLIU Qiang*ZHANG WenmingZHANG ZhijingWANG WeilanZHUANG Su
(College of Animal Science and Technology, Nanjing Agricultural University, Nanjing 210095, China)
The microbial flora diversity and composition in pigs fed different fibrous diets with or without cell wall-degrading enzyme supplementation were compared using terminal restriction fragment length polymorphism analysis technique. Eight growing castrated pigs fitted with a T-shaped cannula at the terminal ileum were randomly assigned to four diets, i.e., wheat bran-based diet (WB), enzyme-supplemented WB, soybean hull-based diet (SH), and enzyme-supplemented SH, according to a 4×4 Latin square design. The experiment lasted for 6 days after 15 days of adaptation in each period. The results showed as follows: 1) the microbial diversity of ileal digesta of pigs fed SH was significantly higher than that of pigs fed WB (P<0.05). 2) The WB diet increased the abundances ofPrevotellaandLactobacillusin ileal digesta (P<0.05), whereas the SH diet increased the abundances ofRuminococcusin ileal digesta and Bacteroides andLachnospirain feces (P<0.05). 3) Adding cell wall-degrading enzyme significantly increased the abundances ofLachnospiraandEubacteriumin ileal digesta andLachnospirain feces (P<0.05), whereas decreased the abundances ofLactobacillus,Prevotellain ileal digesta andLactobacillus,BacteroidesandKlebsiellain feces (P<0.05). In conclusion, dietary fiber significantly increases the abundances of fiber-degrading bacteria, and cell wall-degrading enzyme supplementation selectively alters the microbial diversity and composition.[ChineseJournalofAnimalNutrition, 2016, 28(10):3275-3283]
growing pigs; dietary fiber; cell wall-degrading enzyme; gastrointestinal microbiota; terminal restriction fragment length polymorphism
, associate professor, E-mail: liuayang@njau.edu.cn
10.3969/j.issn.1006-267x.2016.10.031
2016-04-02
国家自然科学基金项目(31172237)
张永婧(1990—),女,天津人,硕士研究生,动物营养与饲料科学专业。E-mail: 1085285725@qq.com
刘强,副教授,硕士生导师,E-mail: liuayang@njau.edu.cn
S816.35
A
1006-267X(2016)10-3275-09
