运用全外显子组测序技术诊断非综合征性耳聋基因变异
2016-11-15刘维强张慧敏余国就孙筱放
刘维强 张慧敏 余国就 孙筱放
·论著·
运用全外显子组测序技术诊断非综合征性耳聋基因变异
刘维强 张慧敏 余国就 孙筱放★
目的评价全外显子组测序(whole-exome sequencing,WES)技术在非综合征性耳聋患者基因变异诊断临床应用的可行性。方法利用Illumina Hiseq2000平台对15例耳聋患者标本进行全外显子捕获测序,重点关注80个耳聋相关基因的序列捕获效率及变异检测情况并运用常规测序验证。结果目标区域(region of interested,ROI)20X以上平均覆盖度为98%;在15份耳聋患者标本中均发现致病性变异,涉及GJB2、SLC26A4、DSPP、TECTA和CHD7基因,其中,无义突变2个,移码突变4个,剪切位点变异1个,其余为错义突变。经常规Sanger测序验证,结果一致。结论全外显子组测序技术可快速可靠诊断非综合征性耳聋患者基因变异。
全外显子组测序;耳聋;变异
耳聋是最常见的人类感觉神经疾病,其在儿童中的发病率可达1/1 000到1/300[1]。超过50%的遗传性耳聋属于感音性耳聋[2],其中的70%为非综合征性耳聋。耳聋是一种遗传异质性非常高的疾病,目前已明确的非综合征和综合征性耳聋基因已超过200个,并且仍有大量遗传性耳聋致病基因尚不明确。虽然耳聋基因检测并不能对治疗产生直接帮助,但其对明确家系致病位点以及产前诊断都起着非常关键的作用,因此对耳聋致病基因的分子诊断有着重要的意义。
我国常见的耳聋致病基因为GJB2、SLC26A4和12SrRNA,常见的检测方法有基因芯片、荧光定量PCR法及Sanger测序等[3]。这些方法的局限是检测位点少,成本高,检测周期长,阳性率低等。随着下一代测序技术(nextgeneration sequencing,NGS)在临床的广泛应用,基于全外显子组和目标序列捕获的NGS方法已开始应用于耳聋基因变异的检测[4-7]。本研究对15份经常规9个热点变异筛查阴性的耳聋患者标本应用全外显子组捕获测序,并设计目标区域重点针对80个耳聋相关基因进行变异分析。
1 材料与方法
1.1 对象
15例病例标本由广州军区广州总医院友好提供。标本经常规9个热点变异筛查阴性。标本采自诊断明确的具有听力障碍的患者,其中男9例,女6例,年龄1~6岁,平均年龄(3±1.4)岁。15例健康人标本作为正常对照。
1.2 试剂和仪器
DNA提取方法采用DNeasy Tissue试剂盒(Qiagen,德国)。DNA打断仪为Covaris S2系统(Covaris,美国),文库建立采用美国Agilent公司Sure Select试剂,文库质检使用Tape-Stations2200仪器检测(Agilent,美国),测序试剂和仪器来自Illumina,美国。Sanger测序使用Applied Biosystems 3500基因分析仪(ThermoFisher,美国)。
1.3 基因组DNA质量评估
使用Nanodrop仪(ThermoFisher,美国)检测DNA浓度及纯度,-20℃保存备用。
1.4 文库构建及测序反应
3μg基因组DNA经Covaris S2系统打断为200~300 bp大小片段,使用美国Agilent公司的SureSelect双向测序试剂进行文库富集。750 ng文库使用美国Agilent公司的SureSelect Human All Exon 50Mb试剂进行目标区域的捕获。捕获的文库经加标签混匀后利用美国Illumina公司的HiSeq2000测序仪90个循环全外显子组测序。
1.5 数据分析
数据经Illumina测序仪自带软件CASAVA(1.8.2版本)拆分得到原始FASTQ文件。原始数据经Trimmimatic软件去掉接头和低质量数据,得到的高质量数据用BWA软件比对到人参考基因组hg19(http://genome.ucsc.edu/)上进行初步分析。原始数据转化成FASTA文件后导入NextGeNe软件(Softgenetics,美国)及生成VCF文件后再导入wannovar(http://wannovar.usc.edu/)网站进行注释分析。
1.6 数据验证
对NextGeNe和wannovar软件发现的致病性变异利用Sanger测序进行验证。对目的基因进行外显子的引物设计,PCR及产物纯化后上Applied Biosystems 3500基因分析仪(ThermoFisher,美国)进行测序,结果采用Mutation Surveyor进行数据分析。
2 结果
2.1 DNA质量评价
所有DNA浓度均介于150 ng/μL~200 ng/μL间,A260/A280比值介于1.8~2.0间,质量合格。
2.2 文库质量评价
文库质量经Tape-Stations仪器检测,片段大小、浓度均符合要求,所建文库片段大小介于315 bp左右。左边峰及最右边峰为标准质量品,大小为25 bp和1 500 bp,中间峰为待检文库片段,平均大小为315 bp,见图1。
2.3 测序数据分析
将原始测序数据经格式转换导入NextGeNe软件进行处理,各标本的测序数据经与基因组参考序列比对得到有效读数介于3兆到6兆。其中80个耳聋基因目标区域的有效测序深度及测序覆盖度如表1所示。测序数据的分布具有较好的随机性,双向测序数据比接近1∶1,不存在偏倚性(图2)。
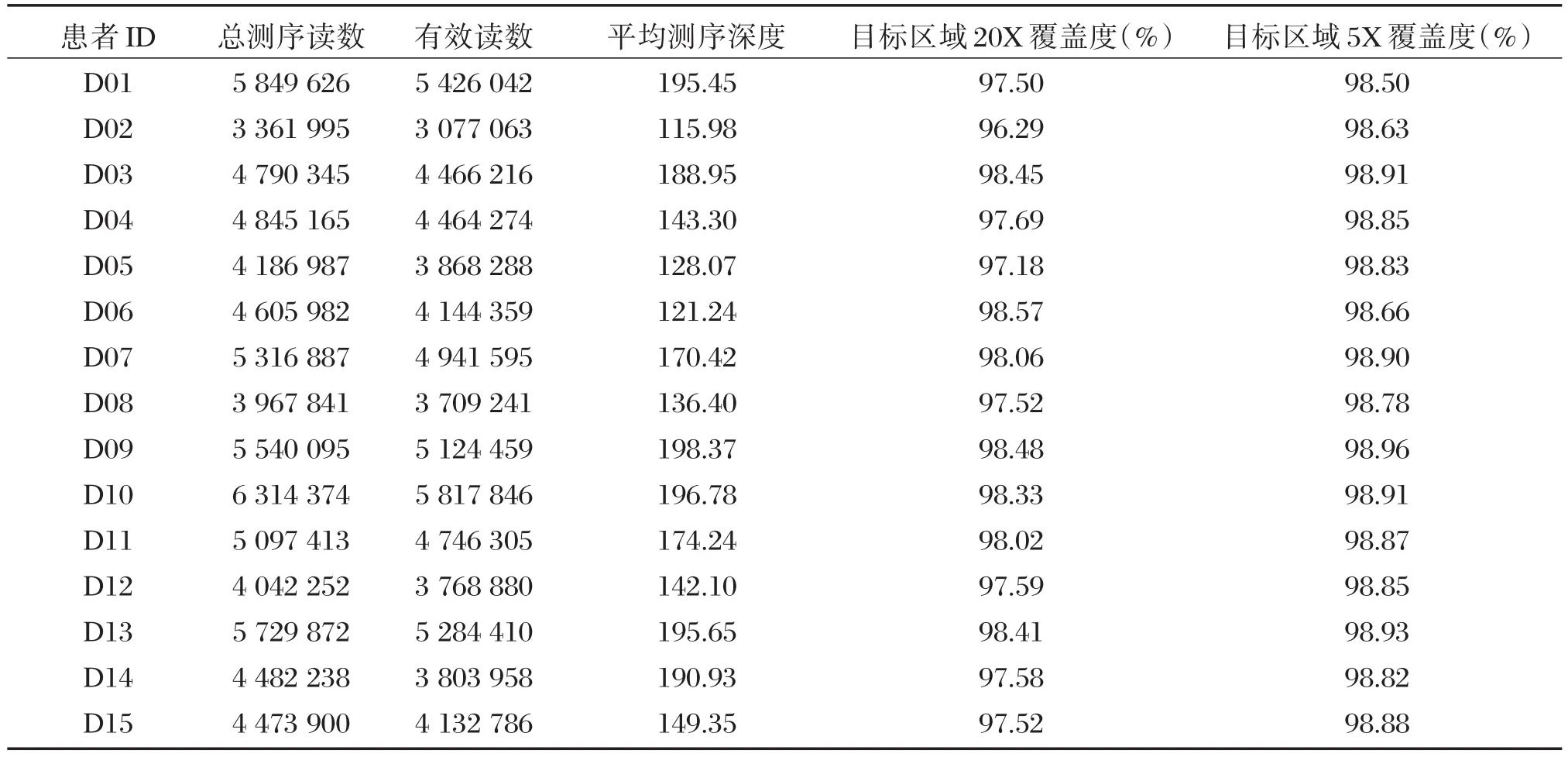
表1 测序数据一揽表Table 1 Statistic of NGS data
2.4 变异检测情况及功能预测
通过软件自动分析及数据过滤,本研究在15份耳聋患者标本中均发现致病性变异,除了涉及GJB2、SLC26A4 2个常见基因外,还在DSPP、TECTA和CHD7 3个基因中发现了变异。在这些变异中含无义突变2个,移码突变4个,剪切位点变异1个,其余为错义突变。变异位点见表2。对变异可能对蛋白功能产生的影响经mutationtaster等在线软件进行预测,结果见表2、图3。
2.5 变异验证
Sanger测序证实全外显子组测序发现的变异结果正确,见图4。
3 讨论
我国常见的耳聋致病基因为GJB2、SLC26A4和12SrRNA,其中9个变异位点是中国人群耳聋基因变异热点,包括GJB2基因的4个变异c.35delG、c.176del16、c.235delC、c.299-300delAT,GJB3基因的一个变异c.538C>T,SLC26A4基因的2个变异位点c.IVS7-2A>G、c.2168A>G和线粒体m tDNA 12SrRNA的2个位点1555A>G、1494C>T。虽然对以上热点变异运用基因芯片等技术可以进行变异的快速筛查,但由于耳聋基因的高度异质性,与耳聋相关的基因目前已超过200个,因此这种热点筛查的方法仍可能导致很大一部分耳聋患者不能明确基因变异情况。
近年来NGS技术已开始应用于遗传性耳聋致病基因的研究[5,8-9]。运用NGS技术,一些新的耳聋致病基因和位点也不断被发现[5,10]。目前运用较多的NGS测序是目标区域捕获测序或在全外显子水平进行变异位点的检测。目标区域捕获技术可重点关注与耳聋等疾病密切相关的十几个或几十个基因,而全外显子组测序则更有助于在所有蛋白编码基因序列及其附近非编码区找到罕见或新发变异。本研究前期对100多份耳聋病例进行了常规变异热点的检测,发现仍有近一半病例未能找到明确的致病变异。对其中15个病例常规检测阴性的标本我们进行了全外显子组测序。虽然从经济成本考虑,目标区域捕获技术相对更有优势,但为了避免遗漏可能的罕见新发变异,本研究尝试运用全外显子组测序技术进行检测。为方便数据分析,本研究设计了80个耳聋相关基因的兴趣区域。
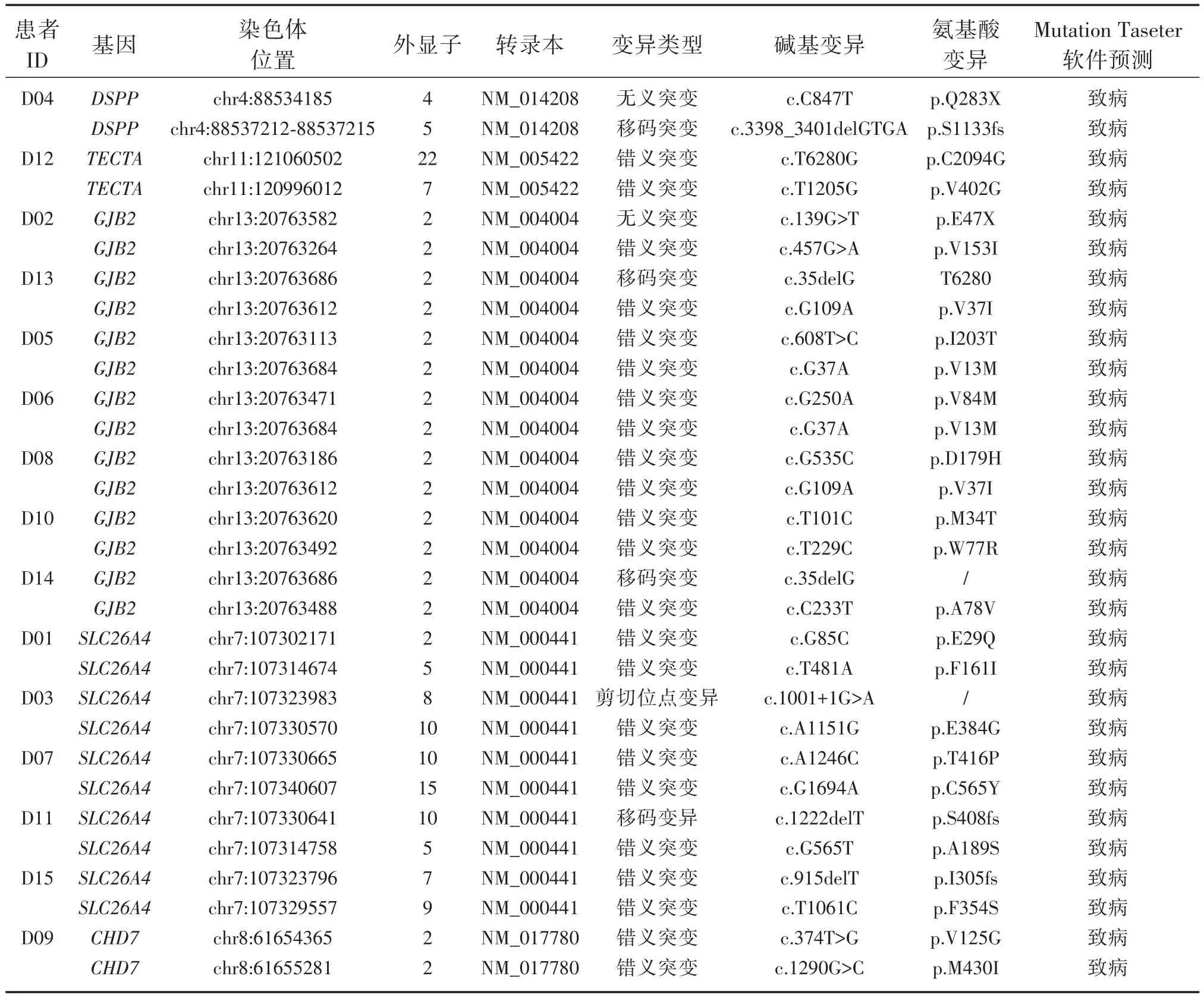
表2 耳聋基因变异检测情况Table 2 Functionalprediction of the identified variants
对全外显子组数据分析是发现变异的关键。通常一份标本的全外显子组测序结果一般可发现2万到5万个变异[11],因此从如此众多的变异中找寻到与耳聋密切相关的变异非常困难。本研究首先对数据进行数据过滤,将单核苷酸多态(single nucleotide polymorphism,SNP)数据库、千人基因组等数据库中已报道的频率大于3%的多态性变异过滤,然后通过查询文献,并重点关注80个耳聋相关基因的兴趣区域进行软件自动查对,再通过生物信息网站如mutationtaster、SIFT和Polyphen-2对变异进行功能预测[12-14]。通过以上方法,本研究在GJB2、SLC26A4、DSPP、TECTA和CHD7等基因发现变异,除无义突变,移码突变等明确导致疾病的变异外,其他大部分变异均为错义突变。由于对错义突变的预测运用不同的预测软件可以有不一致的结果,有些软件认为变异致病而另一些软件则认为是多态性,所以会给数据分析带来一定的困惑。在本研究中,为避免这种情况,我们联合运用前文所述的3种生物功能预测软件来对同一个错义变异进行联合分析。当3种软件均认为是致病或可能致病时才保留下来进行后续分析。另外对变异是否致病我们同时还对位点的保守性进行评估,当变异发生在高度保守区时则格外关注。
在本研究中,我们发现中国人群耳聋基因变异仍是以GJB2、SLC26A4为主。尽管如此,我们也只在2个病例中发现了常见的35delG变异,并且这2个病例如果运用常规方法只能找到一个杂合变异位点,容易导致诊断错误,另外的大部分变异如果用常规方法不能检出,证实常规热点筛查方法的确存在局限性。本研究发现4号病例DSPP基因的2个变异,文献报与DSPP基因变异与遗传性牙本质发育不全相关[15],而此病也可伴有耳聋表型[16],考虑到本病例变异一个是无义变异,另一个是移码突变,而这2个变异均可导致蛋白编码提前终止,因此我们认为这2个杂合变异可能为可能致病性变异,但仍需进行后续的功能验证实验。另外对12号病例我们在TECTA基因发现2个杂合变异,通过检索相关数据库未见明确致病报道,但对其可能影响蛋白的功能预测及保守性分析后我们认为此变异为可能致病性变异。
总之,本研究运用全外显子组测序技术快速诊断出耳聋患者的多个致病变异,证实全外显子组测序技术在耳聋变异诊断中的应用价值。对耳聋致病基因的分子诊断,联合运用热点变异筛查和全外显子或目标区域基因组合测序将有助于发现潜在的致病变异位点。随着NGS在大样本耳聋基因变异检测中的应用,此技术将体现出越发明显的优势。
[1]Mehl AL,Thomson V.The Colorado newborn hearing screening project,1992-1999:on the threshold of effective population-based universal newborn hearing screening[J].Pediatrics,2002,109(1):E7.
[2]NanceWE,Lim BG,Dodson KM.Importance of congenital cytomegalovirus infections as a cause for pre-lingual hearing loss[J].JClin Vird,2006,35(2):221-225.
[3]Lim BG,Clark RH,Kelleher AS,et al.Utility of genetic testing for the detection of late-onset hearing loss in neonates[J].American journal of audiology,2013,22(2):209-215.
[4]Patel K,Giese AP,Grossheim JM,et al.A novel C-term inal CIB2(calcium and integrin binding protein 2)mutation associated with non-syndromic hearing loss in a hispanic fam ily[J].Plos one,2015,10(10):e0133082.
[5]Gao J,Wang Q,Dong C,etal.Whole exome sequencing identified MCM 2 asa novel causative gene for autosomal dominant nonsyndromic deafness in a Chinese fam ily[J].Plosone,2015,10(7):e0133522.
[6]Li-Yang MN,Shen XF,WeiQJ,etal.IVS8+1 DelG,a novel splice site mutation causing DFNA5 deafness in a Chinese family[J].Chinese Medical Journal,2015,128(18):2510-2515.
[7]Chang MY,Kim AR,Kim NK,et al.Identification and clinical implications of novel MYO15A mutations in a non-consanguineous Korean family by targeted exome sequencing[J].Molecules and cells,2015,38(9):781-788.
[8]Atik T,Bademci G,Diaz-Horta O,et al.Wholeexome sequencing and its impact in hereditary hearing loss[J].Genetics research,2015,97:e4.
[9]Ammar-Khodja F,Bonnet C,Dahmani M,et al.Diversity of the causal genes in hearing impaired A lgerian individuals identified by whole exome sequencing[J]. Molecular Genetics&Genomic Medicine,2015,3(3):189-196.
[10]Liu F,Hu J,Xia W,et al.Exome sequencing identifies amutation in EYA 4 as a novel cause of autosomal dominant non-syndromic hearing loss[J].Plos one,2015,10(5):e0126602.
[11]Abecasis GR,Altshuler D,Auton A,et al.A map of human genome variation from population-scale sequencing[J].Nature,2010,467(7319):1061-1073.
[12]Kumar P,Henikoff S,Ng PC.Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm[J].Nature protocols,2009,4(7):1073-1081.
[13]Adzhubei IA,Schmidt S,Peshkin L,et al.A method and server for predicting damaging missense mutations[J].Nature methods,2010,7(4):248-249.
[14]Schwarz JM,Cooper DN,SchuelkeM,etal.Mutation taster2:mutation prediction for the deep-sequencing age[J].Nature methods,2014,11(4):361-362.
[15]Malmgren B,Lindskog S,Elgadi A,et al.Clinical,histopathologic,and genetic investigation in two large families with dentinogenesis imperfecta type II[J].Human genetics,2004,114(5):491-498.
[16]Xiao S,Yu C,Chou X,et al.Dentinogenesis imperfecta 1 with orw ithout progressive hearing loss is associated with distinct mutations in DSPP[J].Nature genetics,2001,27(2):201-204.
Identification of gene variations in patients with nonsyndromic deafness by using whole-exome sequencing method
LIU Weiqiang,ZHANG Huimin,YU Guojiu,SUN Xiaofang★
(Key Laboratory for Reproduction and Genetics of Guangdong Higher Education Institutes,Key Laboratory for Major Obstetric Diseases of Guangdong Province,Third Affiliated Hospital of Guangzhou Medical University,Guangzhou,Guangdong,China,510150)
Objective To assess the efficiency of clinical genetic diagnosis of patients with nonsyndromic deafness by using whole-exome sequencing(WES)method.Methods WESwas performed using the Illumina Hiseq2000 platform in 15 patients with non-syndromic deafness.In this study,we designed a panel containaining 80 deafness genes for variant identification.A ll variants detected by WES were validated by Sanger sequencing.Results The coverage of the region of interest(ROI)over 20X reads was above 98%;pathogenic or likely pathogenic mutations were identified in different genes,such as GJB2,SLC26A4,DSPP,TECTA and CHD7.Among these variations,2 were nonsense mutations,4 were frame shift mutations,1 was a splice site mutation and the remaining were missense mutations.All variants were validated by Sanger sequencing.Conclusion The whole-exome sequencing method has provided a powerful tool to discover rare variations or novel genes in patients with non-syndromic deafness.
Whole-exome sequencing;Deafness;Variants
国家自然科学基金(31171229);广东省医学科研基金(A2015327);广东省科技厅基金(2013B051000087;2014A020212354);广州市科技局基金(201400000004-4;201400000003-4);广州医科大学留学回国人员基金(2013C56)
广东省产科重大疾病重点实验室,广东省普通高校生殖与遗传重点实验室,广州医科大学附属第三医院,广东,广州510150
★通讯作者:孙筱放,E-mail:xiaofangsun@gzhmu.edu.cn
