氨基酸序列特征向量提取方法的探讨
2016-10-31谭生龙
谭生龙

摘要:机器学习算法无法直接对连续的氨基酸序列进行功能注释,将用字符串表示的氨基酸序列转化成用数值表示的特征向量是必要步骤,本文探讨了基于氨基酸序列的特征提取方法,简单探讨了各种方法的优劣,为新的氨基酸序列特征提取方法的研究起到抛砖引玉的作用。
关键词:蛋白质序列;特征向量;特征提取
中图分类号:TP18 文献标识码:A 文章编号:1009-3044(2016)22-0169-02
1 引言
随着测序成本的下降,通过高通量测序获得生物序列的速度正以几何级数增长,如何应对如此快速增长的序列并进行快速的功能注释变得非常必要,一种可行的方法是利用计算机的机器学习方法实现对序列进行快速的功能注释。目前,机器学习算法仅支持对离散特征向量进行学习和分类,而不能对用连续字符串表示的生物序列进行自动分类。生物序列包括核苷酸序列和氨基酸序列,核苷酸序列又可以分为DNA序列和RNA序列,形成DNA序列的字母表为{A,T,C,G},RNA序列的字母表由{A,U,C,G}组成;而蛋白质的氨基酸序字母表为∑={A,C,D,E,F,G,H,I,K,L,M,N,P,Q,R,S,T,V,W,Y},字母表中字符的个数为|∑|=20,氨基酸序列由字母表中的字符生成,本文主要讨论将氨基酸序列转化成离散特征向量的方法及各方法的优缺点。
2 常用生物序列的特征提取策略
1)氨基酸组成成分的特征提取方法
将氨基酸序列转化为特征向量的方法中,最简单的方法是计算字母表中的各个字母在序列中的出现频率。将长度为n的氨基酸序列S表示为:S[1..n]=r1r2..ri..rn,其中ri∈∑,ri是组成氨基酸序列的单个字母,则字母表中的单个字符ri出现的频率为Fi = Ai /n。其中 Ai 为字符ri在序列S中出现的次数。字母表中有20个字符,对不同长度的氨基酸序列,均生成一个有20个分量的特征向量,具体的实现方法见[1]。本方法实现简单,且不同长度的氨基酸序列生成的特征向量长度相同,方便运算和比较,但该方法没有考虑氨基酸序列内部的顺序关系,丢失了序列内部较重要的位置信息。
2)分段伪氨基酸组成成分的特征提取
蛋白质的氨基酸序列具有局部特征,蛋白序列的局部子序列具有功能域的作用。一般把氨基酸序列中具有功能域特征的子片段称为模体(motif),具有功能的氨基酸序列一般由多个功能域构成。由此,将蛋白序列S均分成m个子片段,然后在每个子片段内部计算氨基酸组成成分的特征向量,然后将这m个特征向量拼成一个具有20×m长度的特征向量,具体的实现方法见[2]。
3)基于k-mer频度的特征提取方法
为了考虑氨基酸序列内部的位置关系,以氨基酸序列内部固定长度的小片段为研究对象,计算这些小片段序列的出现频率,称这种小片段为k-mer,k为小片段的长度。当k=1时,本方法即为氨基酸组成成分的特征提取方法。当k=2时,由20种基本氨基酸组成的2-mer有202=400种组合,即包括∑2={AA,AC,AD,…,YY}共400种短片度,生成的特征向量有400个分量。我们可以统计这400种小片段在序列S中的出现频率,由此可将序列S转换为一个400维的向量。计算某个k-mer频率的方法为Fi = Ai /(n-k+1),Ai为单个k-mer在序列S中的出现次数,n为序列S的长度,k为短片段k-mer的长度。当k=3时,蛋白质序列中的3-mer有203=8000种可能,即∑3={AAA,AAC,…,YYY},生成一个8000维的向量,每个分量表示某3-mer在该序列中出现的频率。当k-mer中的长度继续增加时,比如k≥4时,在单条序列S中,k-mer个数太多而单个k-mer在序列S中出现在频率很少,很多k-mer出现0次或者1次,大量由0和1构成的特征向量已经没有意义。比如4-mer生成的特征向量有204=160000个分量,在氨基酸序列S中,多数4-mer仅出现0次,故特征向量中的多数分量是0,因此该向量的维数太高而很少被采用。通过计算同一组蛋白不同k-mer的频率,可以将蛋白的氨基酸序列表示成一组由k-mer频率表示的特征向量,长度不同的氨基酸序列,只要选择相同的k值,其生成的特征向量的维度相同。本方法考虑了序列的相邻关系,但当k增大时,特征向量的维度以几何方式增长,高维特征向量为后续的机器学习算法引入维度灾难,使预测分类性能显著下降。
4)伪氨基酸组成成分
氨基酸组成成分方法没有考虑残基在序列中出现的顺序,而仅仅计算二十种基本氨基酸在序列中的出现频率,该模型忽略了氨基酸残基间的顺序关系;k-mer频度的特征提取方法仅考虑了氨基酸残基的局部顺序关系,当k增加时,向量维度迅速升高;这两种模型在将氨基酸序列转化成特征向量方面丢失了较多的氨基酸残基间的顺序关系。为了更完整的表示序列的位置信息,Chou等人提出了伪氨基酸组成模型[3, 4]PAAC(Pseudo Amino Acid Composition Model),在该模型中,伪氨基酸组成模型保留了氨基酸组成成分的特征,并通过扩展特征向量来表示位置信息。故伪氨基酸组成成分的特征向量表示为:
PAAC =(x1,x2,…,xi,…,x20,x20+1,…,x20+λ) (1)
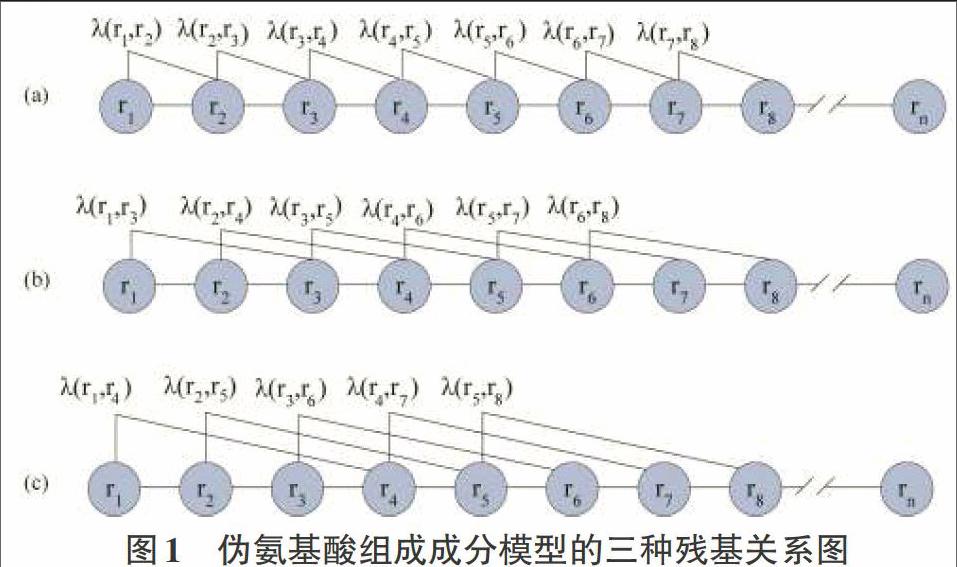
在PAAC中,前20个分量x1,..,x20表示20个基本氨基酸的出现频率,而分量x20+1,…,x20+λ部分表示了氨基酸序列中残基的位置信息。残基间的位置信息能通过如图1所示的残基间的相关关系来描述。图1中的(a)描述残基间的第一层关系,即分量x20+1,(b)和(c)分别描述了残基间的第二层和第三层关系,即分量x20+2和x20+3,层数λ可由用户指定,但λ应该小于蛋白序列的长度n。
图1中氨基酸序列的残基之间的相关关系可以用下面的公式计算:
…
在这里x20+1表示氨基酸序列间的第一层关系,x20+2表示氨基酸序列的第二层关系,x20+λ表示氨基酸序列的第λ层关系,λ是一个输入参数;n表示氨基酸序列的长度,λ(ri,rj),表示两个基本氨基酸残基之间的关系。通常,λ(ri,rj)是用两基本氨基酸的理化性质、空间结构改变或者序列间的转化等数量关系来描述。
5)组合的特征向量生成方法
在应用这些特征提取方法将蛋白的氨基酸序列转化成特征向量的过程中,可同时使用几种特征向量提取方法,将由不同特征提取方法生成的特征向量进行组合,生成组合的特征向量,实践证明这种组合特征向量能更好地表示序列S的内部信息,这种组合特征向量能较好的提升机器学习算法的预测性能。在应用这些方法的过程中,经常遇到因为特征向量维度过高引发的维度灾难问题,一般采用主成分分析、F-Score和二项分布[5]等多种方式对特征向量进行降维。
3 总结
蛋白序列的特征提取技术直接决定机器学习分类模型的预测性能和预测结果的准确性。高效的特征提取技术能将连续的氨基酸序列快速转化为离散的特征向量,且更好展现了序列的内部隐藏信息。在生物序列数据快速增长的新形势下,研究高效特征提取技术已经变得非常必要,本文对目前的氨基酸序列的特征提取技术进行了综述,希望能对高效特征提取方法的研究起到抛砖引玉的作用。
参考文献:
[1] Nakashima H,Nishikawa K.Discrimination of intracellular and extracellular proteins using amino acid composition andresidue—pair frequencies.J Mol Biol,1994,238(1):54-61.
[2] 杨会芳,程咏梅,张绍武,等. 基于一种新的特征提取方法 分段伪氨基酸组成成分预测蛋白质亚细胞定位 生物物理学报,2008,24(33):232-238.
[3] K. C. Chou, Prediction of protein cellular attributes using pseudo-amino acid composition, Proteins: Structure, Function, and Bioinformatics, 2001(43):246-255.
[4] K.-C. Chou, Some remarks on protein attribute prediction and pseudo amino acid composition, Journal of Theoretical Biology, 2011(273):236-247.
[5] Lin H, Deng E, Ding H. iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Research, 2014, 42(21), 12961-12972. doi:10.1093/nar/gku1019.
