固相萃取-气相色谱-负化学源质谱法检测水果和蔬菜中的毒杀芬
2016-10-25张慧丽王建华姜瑛王颖辛学谦汤志旭
张慧丽,王建华,姜瑛,王颖,辛学谦,汤志旭
(1. 中国海洋大学,山东青岛 266100; 2.山东出入境检验检疫局检验检疫技术中心,山东青岛 266002;3.山东省海洋生物研究院,山东青岛 266104)
固相萃取-气相色谱-负化学源质谱法检测水果和蔬菜中的毒杀芬
张慧丽1,2,王建华2,姜瑛2,王颖3,辛学谦1,2,汤志旭2
(1. 中国海洋大学,山东青岛 266100; 2.山东出入境检验检疫局检验检疫技术中心,山东青岛 266002;3.山东省海洋生物研究院,山东青岛 266104)
采用改进的分散固相萃取(QuEchERs)法对样品进行前处理,建立水果和蔬菜中毒杀芬残留的气相色谱-负化学电离源质谱(GC-NCI-MS)检测方法。样品中的毒杀芬由正己烷提取,经吸附剂PSA+GCB净化,在GC-NCIMS的选择离子扫描模式下进样分析。毒杀芬的色谱保留时间在12.5~18.0 min区间内,采用面积归一化法积分,外标法定量。毒杀芬质量浓度在0.050~2.000 mg/L范围内与色谱峰面积呈良好的线性关系,相关系数r=0.999 1。分别以蓝莓、黄桃、菠菜为基质,在0.025,0.050 mg/kg添加水平下,毒杀芬的回收率为107.2%~118.1%,测定结果的相对标准偏差为5.5%~8.8%(n=6),定量限为0.025 mg/kg。该方法检测快速,适用于水果和蔬菜中毒杀芬残留的日常检测。
蔬菜;水果;气相色谱-负化学源质谱法;毒杀芬;检测
毒杀芬是多氯化合物组成的混合物,平均元素组成为C10H10Cl8,氯含量为67%~69%[1-3]。毒杀芬作为有机氯类杀虫剂具有广谱杀虫活性,自20世纪70年代美国环保署颁布滴滴涕(DDT)禁令后,毒杀芬已取代DDT并成为主要的农业杀虫剂。毒杀芬容易在富含脂质的生物组织中蓄积,已证实在动物和人的母乳中被发现[4-5];也有文献报道毒杀芬对鱼和啮齿类动物具有剧毒及雌激素的作用,可作为一种内分泌干扰物[6],甚至被归类为致癌物质[7]。毒杀芬的持久性、生物蓄积性、内在毒性、远距离传输的稳定性,使其成为国际公认的有机氯污染物之一[8],因此自20世纪90年代后相继被各国禁止生产。意大利自1985年开始禁止使用毒杀芬;美国1990年开始全面禁止使用毒杀芬;2002年,毒杀芬被列为《斯德哥尔摩公约》中12种首要控制的可持续有机污染物之一;我国自2002年6月禁止在果树上使用毒杀芬。毒杀芬理论上有32 768个同系物,目前通过二维气相色谱发现了约1 000个同类物[9]。在土壤、水体、底泥、鱼和海洋哺乳动物体内均检测到毒杀芬[10-12]。目前许多国家和组织规定其在食品中的最大残留限量,如我国GB 2763-2014[13]规定了水果和蔬菜中毒杀芬的残留限量为0.05 mg/kg;德国规定毒杀芬在鱼中的最大残留限量为0.1 mg/kg (以P26,P50,P62三种毒杀芬单体总残留量计)[14]。
目前关于毒杀芬在食品中残留的检测报道不多。Attard Barbini等[7]采用GC-MS的选择离子(SIM)模式分析鱼肉中毒杀芬残留;Bruno Veyrand等[15]采用气相色谱-高分辨质谱(GC-EI-HRMS)方法检测了鱼肝油中9种毒杀芬同系物,样品经加速溶剂萃取后,采用硅胶柱和弗罗里硅土柱两步净化;廖且根等[16]研究了用气相色谱-质谱法测定动物源性食品中毒杀芬的残留量,样品经乙腈提取,弗罗里硅土净化,采用选择离子检测模式;劳文剑[17]建立了气相色谱-负化学电离源质谱法测定沉积物和鱼肉中毒杀芬的8个同类物及其总量;刘志斌等[18]使用索式提取系统(Soxtec)提取,由硅胶柱和氧化铝柱净化,利用同位素稀释-高分辨气相色谱/高分辨双聚焦磁式质谱(ID-HRGC/HRMS)技术对食品样品中3种指示性毒杀芬单体P26,P50,P62 进行了定量、定性分析。
目前毒杀芬残留检测的样品基质以鱼肉、土壤等居多,尚未见水果和蔬菜中毒杀芬残留测定方法的报道。基于文献报道,笔者对提取溶剂和分散固相萃取吸附材料进行优化,采用改进的分散固相萃取(QuEchERs)法进行样品前处理,用GC-NCI-MS检测水果和蔬菜中毒杀芬总量的残留。该方法提高了萃取效率、减少了溶剂用量,提高了电负性强的毒杀芬物质的响应值和选择性、减少了基质干扰。
1 实验部分
1.1 主要仪器与试剂
气相色谱仪:7890型,配备5975C质谱检测器,美国Aglient公司;
气相色谱仪:6890N型,配备ECD检测器,美国Aglient公司;
旋涡振荡器:MS3 Basic型,德国IKA公司;
水平振荡器:HS501型,德国IKA公司;
均质器:T25 Basic型,德国IKA公司;
台式冷冻离心机:3-18K型,美国Sigma公司;
电子天平:PL303型,瑞士Mettler Toledo公司;
毒杀芬标准样品:纯度为99.0%,德国Dr. Ehrenstorfer公司;
PCB180标准样品:纯度为99.0%,德国Dr. Ehrenstorfer公司;
乙二胺-N-丙基硅烷(PSA)净化剂、石墨化炭黑净化剂:博纳艾杰尔科技公司;
正己烷、丙酮、乙腈、甲醇:色谱纯,市售。
1.2 溶液的配制
毒杀芬标准储备溶液:1 000 mg/L,准确称取0.010 0 g毒杀芬标准品,用正己烷定容至10 mL,于4℃下保存备用;
毒杀芬标准工作溶液:用正己烷将毒杀芬标准储备溶液逐级稀释,配制所需浓度的标准工作溶液;
基质匹配标准溶液:将空白样品按照样品前处理步骤提取、净化,取2 mL净化液吹干后,加入0.4 mL的毒杀芬标准工作溶液。
1.3 样品前处理
1.3.1 提取
称取打碎均匀的蓝莓样品10 g于50 mL离心管中,准确加入10 mL正己烷溶液,手动振摇1 min,在均质机上均质后,加入固相萃取盐包(含4 g无水硫酸镁、1 g 氯化钠、1 g柠檬酸钠和0.5 g柠檬酸二钠盐1.5水合物),剧烈振荡30 s,在水平振荡器上振荡20 min,以8 000 r/min离心5 min后待净化。
1.3.2 净化
移取正己烷提取上清液5 mL转移至事先装 有900 mg硫 酸 镁、150 mg C18和15 mg Bulk Carbograph吸附剂的离心管中,涡旋5 min,以8 000 r/min离心5 min后,准确移取2 mL净化液于氮气下吹干,加入0.4 mL正己烷定容,过0.22 μm有机滤膜后,进样分析。
环境检测数据包括水和废水监测,空气与废气监测,土壤、底泥和固废监测,噪声和振动检测,以及生物检测等项目,涉及水环境(地下水、地表水)、空气环境及声环境等环境要素,还涉及工业废水、工业废气、厂界噪声、生活污水及社会生活噪声等各类污染源.依据国家标准分析方法中的100多项污染物计算方法和实验室的检测原始记录,开发污染物分析结果计算的模块,实现结果计算自动化.
1.4 仪器工作条件
1.4.1 气相色谱条件
色谱柱:HP-5MS毛细管柱(30 m×0.25 mm,0.25 μm);载气:氦气(纯度大于99.999%),流量为1.0 mL/min;升温程序:90℃保持5 min,以15℃/min升至310℃,保持7 min,总运行时间为26.67 min;进样口温度:280℃;进样方式:不分流进样;进样体积:1.0 μL。
1.4.2 质谱条件
负化学电离源(NCI源);反应气:甲烷;传输线温度:280℃;电离源和四级杆温度:150℃;溶剂延迟时间:8.0 min;选择离子监测模式(SIM)。
2 结果与讨论
2.1 检测模式的选择
有文献报道采用气相色谱电子捕获检测器(GC-ECD)检测毒杀芬残留[19-20]。实验考察了添加0.050 mg/kg毒杀芬的菠菜样品在GC-ECD上的岀峰情况。结果发现,在毒杀芬岀峰时间段干扰峰较多、选择性差,会导致毒杀芬定量不准确;采用气相色谱-负化学电离源质谱(GC-NCI-MS)进行分析,检测毒杀芬的灵敏度是GC-ECD的4倍,且分离较好,减少了基质干扰,因此选择GC-NCI-MS进行检测。
2.2 质谱条件确定
采用全扫描(Fulscan)模式对毒杀芬标准溶液进行全扫描,12.5~18.0 min区间为毒杀芬的色谱峰,峰宽5.5 min内共有的碎片离子为m/z 35,m/z 37,m/z 70,m/z 71,m/z 72和m/z 73,其中m/z 35的响应值最大,因此选择m/z 35为毒杀芬的定量离子,其余离子为定性离子。毒杀芬在全扫描模式下的质谱图见图1。
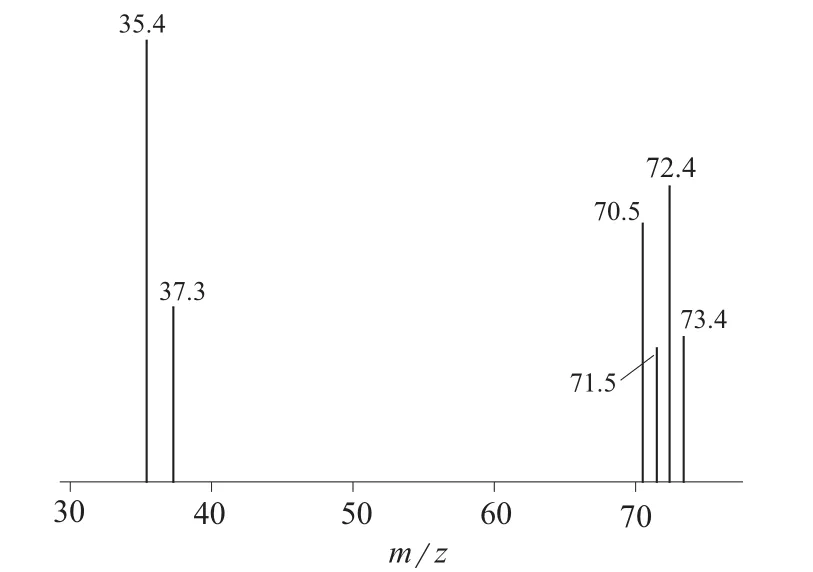
图1 毒杀芬在全扫描模式下的质谱图
2.3 提取试剂的选择
考察了乙腈、正己烷、丙酮-正己烷(体积比为1∶1) 3种溶剂对毒杀芬的提取效率,采用这3种溶剂对添加毒杀芬标准溶液(0.050 mg/L)的蓝莓样品按照1.3方法进行提取、净化,然后进行测定。结果表明,采用乙腈、正己烷、丙酮-正己烷为提取试剂,毒杀芬回收率分别为78.9%,102.5%,37.3%;乙腈提取液颜色为深紫色,净化液颜色为浅黄色;丙酮-正己烷提取液颜色为浅紫色,净化液颜色为微黄色;正己烷提取液颜色为微紫色,净化液为无色清澈透明。用乙腈或丙酮-正己烷提取时,在20 min处有响应较强的杂质峰,而用正己烷提取时杂峰较少。另外在乙腈、正己烷、丙酮-正己烷提取溶剂下,毒杀芬在蓝莓中的基质效应分别为102.8%,115.6%,129.1%(基质效应计算方法见2.6节),采用正己烷和乙腈作为提取溶剂时,毒杀芬的基质效应较弱,而正己烷的提取效率较乙腈高,因此选择正己烷作为提取溶剂。
2.4 净化方法的选择
分散固相萃取常用的吸附剂有PSA,C18,GCB等,以加标菠菜样品的净化液颜色、毒杀芬的回收率和基质效应为评价指标,考察PSA+GCB,PSA+C18,C183种吸附剂类型的净化效果。结果显示,上述3种吸附剂组合,毒杀芬的回收率分别为106.7%,95.5%,92.9%;基质效应分别为115.6%,114.1%,119.6%,均表现为较弱的基质增强效应;从净化液颜色来看,采用PSA+C18,C18净化其净化液颜色均为浅黄色,而采用PSA+GCB净化其净化液为无色且较清澈,表明GCB能够较好地吸附色素类物质。为了进一步比较净化液中的杂质数量以及对毒杀芬的干扰,利用GC-(EI)-MS对3种净化液进行全扫描,结果表明,在毒杀芬12.5~18 min的时间区间内,采用PSA+GCB净化得到的净化液色谱图中杂质峰较少,对毒杀芬的干扰较小,因此选择PSA+GCB作为分散固相萃取吸附剂。
2.5 定量方法的选择
文献[13,15]以多氯联苯作为毒杀芬的内标物,采用内标法定量。分别采用毒杀芬标准加入和PCB180标准加入计算加标黄桃样品中毒杀芬和PCB180的回收率,添加水平为0.025 mg/kg,共6个平行样品。结果表明,6次测定毒杀芬的回收率分别为118.2%,125.5%,120.8%,109.7%,120.4%,106.2%;PCB180的回收率分别为95.2%,92.8%,94.5%,89.5%,98.3%,86.1%。两者的回收率偏差在17.0%~22.9%范围内,结果表明以PCB180为内标的内标法对毒杀芬进行定量不够准确。由于毒杀芬在本实验蓝莓、黄桃、菠菜3种代表性基质中均表现为较弱的基质效应,因此综合考虑,采用外标法进行定量。
2.6 基质效应
基质效应按下式计算:ME=(基质匹配标准溶液的面积/溶剂标准溶液的面积)×100%,经计算在蓝莓基质中毒杀芬的基质效应ME值为115.6%,在黄桃基质中为114.3%,在菠菜基质中为112.4%,说明在蓝莓、黄桃和菠菜中均表现为较弱的基质抑制/增强效应,可忽略不计。
2.7 工作曲线方程与定量限
按1.2配制基质匹配标准溶液的方法配制毒杀芬质量浓度分别为0.050,0.125,0.250,0.500,1.000,2.000 mg/L的系列标准工作溶液,分别进样分析。以毒杀芬质量浓度(X,μg/L)为横坐标,以毒杀芬色谱峰面积(Y )为纵坐标,绘制标准工作曲线;以10倍信噪比确定毒杀芬定量限(LOQ)。毒杀芬的线性方程、相关系数、定量限见表1。
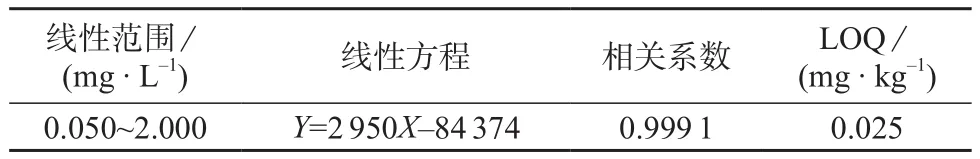
表1 线性方程、相关系数与定量限
2.8 加标回收与精密度试验
在空白蓝莓、黄桃和菠菜样品中添加毒杀芬标准工作溶液进行加标回收试验,添加水平分别为0.025,0.050 mg/kg,每个水平做6个平行样品。毒杀芬的平均回收率和测定结果的相对标准偏差见表1。由表1可知,毒杀芬在3种样品中的平均回收率为107.2%~118.1%,测定结果的相对标准偏差为5.5%~8.8%,所建方法的精密度和准确度满足农药残留分析要求。毒杀芬标准溶液、菠菜空白样品以及加标菠菜样品的总离子流色谱图分别见图2、图3、图4。
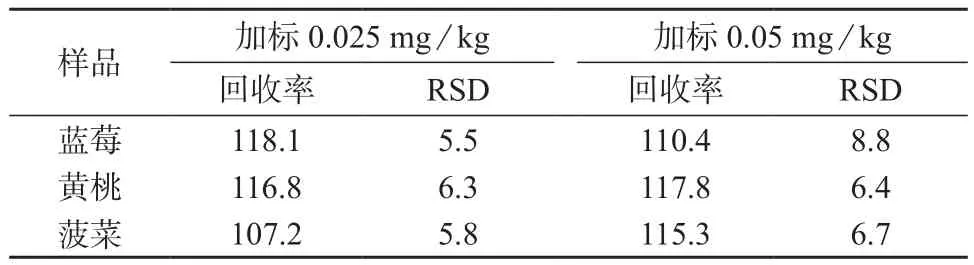
表2 回收率与精密度试验结果(n=6) %
图2 毒杀芬标准溶液的总离子流色谱图
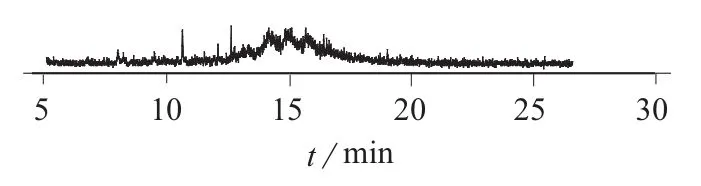
图3 菠菜空白样品的总离子流色谱图
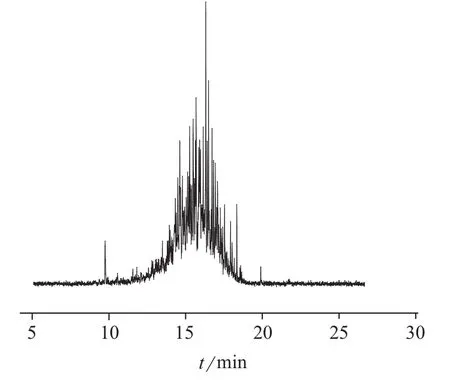
图4 加标菠菜样品的总离子流色谱图
3 结语
通过优化提取试剂和吸附剂材料及对检测仪器进行选择,建立了固相萃取-气相色谱-负化学电离源质谱法分析水果、蔬菜中毒杀芬的含量,该方法试剂用量少,萃取效率高,基质干扰小,选择性好;采用外标法定量,方法的精密度、准确度满足农药残留分析要求,可用于水果、蔬菜中毒杀芬的日常检测。
[1] Holmstead R L,Khalifa S,Casida J E. Toxaphene composition analyzed by combined gas chromatography-chemical ionization mass spectrometry[J]. Journal of Agricultural and Food Chemistry,1974,22(6): 939-944.
[2] Casida J E,Saleh M A. Toxaphene composition and toxicology[M]. New York: US Environmental Protection Agency, Office of Research and Development, Health Effects Research Laboratory,1978: 1-47.
[3] Pollock G A,Kilgore W W. Toxaphene[M]. New York: Springer New York, 1978: 87-140.
[4] Veyrand B,Venisseau A,Marchand P,et al. Determination of toxaphene specific congeners in fish liver oil and feedingstuff using gas chromatography coupled to high resolution mass spectrometry[J]. Journal of Chromatography B,2008,865(1): 121-126.
[5] De Boer J,Wester P G. Determination of toxaphene in human milk from Nicaragua and in fish and marine mammals from the northeastern Atlantic and the North Sea[J]. Chemosphere,1993,27(10): 1 879-1 890.
[6] Saleh M A. Toxaphene: Chemistry,biochemistry,toxicity and environmental fate[M]. New York: Springer New York, 1991:1-85.
[7] Barbini D A,Stefanelli P,Girolimetti S,et al. Determination of Toxaphene Residues in Fish Foodstuff by GC-MS[J]. Bulletin of Environmental Contamination and Toxicology,2007,79(2): 226-230.
[8] Gouteux B,Lebeuf M,Trottier S,et al. Analysis of six relevant toxaphene congeners in biological samples using ion trap MS/MS[J]. Chemosphere,2002,49(2): 183-191.
[9] Korytar P,Van Stee L L P,Leonards P E G,et al. Attempt to unravel the composition of toxaphene by comprehensive twodimensional gas chromatography with selective detection[J]. Journal of Chromatography A,2003,994(1): 179-189.
[10] Musial C J,Uthe J F. Widespread occurrence of the pesticide toxaphene in Canadian east coast marine fish[J]. International Journal of Environmental Analytical Chemistry,1983,14(2):117-126.
[11] De Geus H J,Besselink H,Brouwer A,et al. Environmental occurrence,analysis,and toxicology of toxaphene compounds[J] . Environmental Health Perspectives,1999,107(Suppl 1):115-144.
[12] Wong F,Alegria H A,Bidleman T F. Organochlorine pesticides in soils of Mexico and the potential for soil-air exchange[J]. Environmental Pollution,2010,158(3): 749-755.
[13] GB 2763-2014 食品中农药最大残留限量[S].
[14] Fromberg A,Cederberg T,Hilbert G,et al. Levels of toxaphene congeners in fish from Danish waters[J]. Chemosphere,2000,40(9): 1 227-1 232.
[15] Veyrand B,Venisseau A,Marchand P,et al. Determination of toxaphene specific congeners in fish liver oil and feedingstuff using gas chromatography coupled to high resolution mass spectrometry[J]. Journal of Chromatography B,2008,865(1):121-126.
[16] 廖且根,戴廷灿,陈光宇,等.气相色谱-质谱法应用于动物源性食品中毒杀芬残留测定[J].现代科学仪器,2010(6):105-109.
[17] 劳文剑.气相色谱-负化学电离质谱法测定沉积物和鱼肉中毒杀芬的8 个同类物及其总含量[J].色谱,2013,31(7): 667-673.
[18] 刘志斌,张建清,蒋友胜,等.同位素稀释-气相色谱/高分辨质谱法测定食品中的指示性毒杀芬[J].食品安全质量检测学报,2014(2): 475-484.
[19] Andrews P,Headrick K,Pilon J C,et al. An interlaboratory round robin study on the analysis of toxaphene in a cod liver oil standard reference material[J]. Chemosphere,1995,31(11): 4 393-4 402.
[20] YC/T 180-2004 烟草及烟草制品 毒杀芬农药残留量的测定 气相色谱法[S].
Detection of Toxaphene in Fruits and Vegatables by Gas Chromatography-Negative Ion Mass Spectrometry With Dispersive Solid Phase Extraction
Zhang Huili1,2, Wang Jianhua2, Jiang Ying2, Wang Ying3, Xin Xueqian1,2, Tang Zhixu2
(1. Ocean University of China, Qingdao 266100, China;2. Inspection and Quarantine Technical Center, Shandong Entry-Exit Inspection and Quarantine Bureau, Qingdao 266002, China;3. Marine Biology Institute of Shandong Province,Qingdao 266104, China)
A method was developed for the determination of toxaphene residues in fruits and vegetables by gas chromatography-negative ion mass spectrometry (GC-NCI-MS) with modified QuEChERS sample preparation technique. The samples were extracted by hexane, and cleaned up by the dispersive solid-phase extraction sorbent PSA+GCB,finally analyzed by GC-NCI-MS with selected ion monitoring mode. The chromatographic retention time of toxaphene was from 12.5 to 18.0 min, and quantification was performed for toxaphene by using external standard method. The mass concentration of toxaphene was linear with chromatographic peak area in the range of 0.050-2.000 mg/L, the correlation coefficient was 0.999 1. Blueberry, yellow peach and spinach were taken as matrix, toxaphene recovery was 107.2%-118.1%, and the relative standard deviation was 5.5%-8.8%(n=6) at spiking levels of 0.025, 0.050 mg/kg. The limit of quantification was 0.025 mg/kg. This method is rapid, and it can be used for routine determination of toxaphene residues in fruits and vegetables.
vegatable; fruit; gas chromatography-negative ion mass spectrometry; toxaphene; detection
O657.7
A
1008-6145(2016)05-0053-05
10.3969/j.issn.1008-6145.2016.05.014
联系人:王建华;E-mail: whywrs9@163.com
2016-07-12
