HMGB1与小胶质细胞在阿尔茨海默病发生发展中的研究进展
2016-10-20孔微微刘绪华
孔微微 刘绪华 曹 红 李 军
温州医科大学附属第二医院麻醉科,浙江温州325027
HMGB1与小胶质细胞在阿尔茨海默病发生发展中的研究进展
孔微微刘绪华曹红李军▲
温州医科大学附属第二医院麻醉科,浙江温州325027
高迁移率族蛋白B1(High mobility group box protein-1,HMGB1)和小胶质细胞在阿尔茨海默病(Alzheimer's disease,AD)疾病的发生发展中起了重要的作用。HMGB1不但参与了其神经炎症的过程,并能抑制小胶质细胞对Aβ清除。通过受体结合激活了NF-κB轴信号通路的级联反应,启动基因转录和表达,释放大量炎症介质和转录产物,加重了AD的炎症反应,但有研究表明不同转录因子有不同作用,如P50敲除具有保护作用,但P65激活也具有保护作用。HMGB1通过紧密结合Aβ42,使Aβ42处于稳定的寡聚体状态,减弱小胶质细胞对Aβ42清除的能力。Aβ可直接与小胶质细胞上TLR结合,尤其是TLR2、TLR4,可以促进小胶质细胞分泌各种细胞因子和趋化因子,正反馈促进小胶质细胞的激活以及对Aβ的清除。HMGB1和小胶质细胞对Aβ清除之间失衡也是AD发生及进展的重要原因之一,因此HMGB1可能成为治疗AD的一种新的靶标。
阿尔兹海默病;β-淀粉样蛋白;小胶质细胞;高迁移率族蛋白B1
[Abstract]High mobility group protein B1(HMGB1)and microglia play an important role in Alzheimer's disease(AD)development.HMGB1 not only involve in the process of the inflammation of the nerve,and can inhibit the removal of Aβ.With combining the receptor,HMGB1 can activate a cascade of NF-κB signal pathway,promote gene transcription and expression,release lots of inflammatory mediators and transcription products,aggravate the inflammatory reaction in AD.But some studies have shown that different transcription factors have different effects,such as P50 knocked has a protective effect,but p65 activated also have a protective effect.HMGB1 tight binding of Aβ42,which make the Aβ42 in a stable state of the oligomer,weakening the ability of remove the Aβ42 in microglia.Aβ can be directly combined with TLR in microglia,especially TLR4 and TLR2,which can promote the secretion of various cytokines and chemokines,and positive feedback to promote the activation of the removal of Aβ in microglia.The imbalance between HMGB1 and microglia in Aβ clearance is one of the important reasons for the occurrence and progression of AD.Therefore,HMGB1 may be a novel therapeutic target for the treatment of AD.
[Key words]Alzheimer's disease;Beta amyloid;Microglia;High mobility group box protein-1
阿尔茨海默病(Alzheimer's disease,AD)是引起老年性痴呆的最常见原因之一,预计患病率将在2050年增加约50%[1],AD已严重影响老年人的生活质量。AD是一种中枢神经系统疾病,具有退行性、致死性、进行性发展的特点,临床表现为认知障碍和记忆功能紊乱[2],自理能力进行性减退,并伴有行为障碍和各种精神神经症状。但是迄今AD的病因和发病机制仍不清楚。
AD的一个病理学特征是细胞外β-淀粉样蛋白(amyloid-β,Aβ)的沉积并形成老年斑(senile phques,SP),激活的小胶质细胞与其有关[3]。随着神经原纤维缠结和神经元的损失,AD的发病机制包括氧化损伤、突触变性、炎症反应和神经元死亡。根据一些临床试验的报道[5],对AD小鼠模型进行免疫疗法中发现小胶质细胞对Aβ的清除起到了一定的作用。且在AD典型病例的缺血皮质中发现了活性小胶质细胞的大量积聚和老年斑的减少[6]。因此,小胶质细胞对Aβ的清除作用被认为是脑内Aβ清除机制之一[7]。有相关报道指出高迁移率族蛋白B1(high mobility group box protein-1,HMGB1)与老年斑有关,并且在AD大脑中HMGB1的含量升高[8]时,可抑制小胶质细胞对Aβ的清除,从而参与了AD的病理过程[9]。因此,小胶质细胞对Aβ的吞噬功能失调可能由细胞外Aβ板块中积聚的HMGB1引起,这可能对AD的病程进展起到重要的作用。
1 HMGB1
1.1 HMGB1的结构及生物学功能
HMGB1是坏死细胞所释放的的非组蛋白染色体结合蛋白,具有两个同源结构域,A-box和B-box,B-box是引起炎性反应的功能结构域和晚期糖化终产物受体(RAGE)结合域,而A-box对B-box有一定的拮抗作用[10]。HMGB1作为炎症刺激激活的单核细胞和内皮细胞分泌释放大量炎性细胞因子,如TNF-α、IL-1、IL-8、MIP-1等[11-14],而炎症细胞因子又反过来促进HMGB1的分泌,形成了一个正反馈环路,使炎症反应不断得以放大、加重,这种正反馈效应对炎性反应的维持起到了相当重要的作用,因而促进了慢性炎症的发生[15]。
HMGB1存在于真核细胞的细胞质和细胞核中,还可在两者之间自由穿梭,且呈细胞周期依赖性。除了参与DNA复制、重组、转录、修复外,还参与了李炎症反应、细胞分化、细胞增生、组织再生、肿瘤发展等过程[16,17]。
1.2 HMGB1的受体及相关信号转导通路
HMGB1的识别及跨膜信号转导需要一系列分子的参与(图1),其受体主要是晚期糖基化终产物(RAGE)和Toll样受体等[18]。
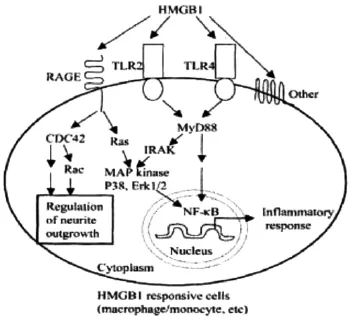
图1 HMGBl与其受体的作用途径
RAGE(receptor for advanced glycation end products)是一种多配体受体,属于免疫球蛋白超家族,广泛存在于体内多种细胞表面[19],而HMGB1和淀粉样蛋白Aβ也是其的主要受体之一。研究表明[20],HMGB1与Aβ结合后能有效抑制小胶质细胞对Aβ的清除及降解。增多的Aβ能与RAGE结合也启动了RAGE/ NF-κB轴的级联反应。同时,HMGB1与RAGE结合后产生大量氧自由基,激活IκB激酶,使IκB磷酸化并解离,使NF-κB具有活性。NF-κB又与TNF-α、IL-1、IL-6等多种细胞因子的基因启动子特异性结合,促进基因转录和表达,其靶基因产物又反过来激活NF-κB,诱发级联放大效应。因此,NF-κB通过RAGE的过度表达形成一个正反馈循环[21]。有研究表明[22],HMGB-1在P50敲除的小鼠具有保护效应,且表明HMGB-1结合后引起RAGE/NF-κB轴NF-κB信号传导通路具有正向调节线。相反,NF-κB信号通路,特别是p65激活,也参与了破坏神经元效应[23]。这个现象可能是由于NF-κB蛋白是一个转录因子复合家庭(p50、p65,P52,c-Rel、RelB)成员,可以结合形成异源二聚体和不同组成的同型二聚体,从而可以在一个给定的细胞类型和产生不同的甚至相反的功能激活的差异[21,22]。
1.3 Toll样受体(TLR)及相关通路
TLR(Toll-like receptor)是一类天然免疫受体,通过对不同外源性的配体结合后,并引发一系列不同途径信号转导,进而导致大量炎性介质(如IL-12、IL-8和IL-23)的释放,在非特异性免疫中起着重要作用。目前发现与HMGB1相关的TLR主要是TLR2、TLR4、TLR9[24],其中TLR4是内毒素的主要受体,而TLR2对革兰阳性菌的组分和真菌起反应[25]。TLR2作为血源性病原体及炎性细胞因子的靶向目标。TLR2存在于动脉内皮细胞以及其他细胞中的造血谱系。TLR2可以介导不同的信号路径所导致NF-κB的活化和核易位。TLR4是介导内毒素/脂多糖(lipopolysaccharide,LPS)应答的最主要受体,主要表达在参与宿主防御功能的细胞上,介导病原相关分子模式的识别,TLR4也有报道在神经退行性疾病中起作用,包括阿尔茨海默病、肌萎缩侧索硬化症、多发性硬化症和帕金森病[26]。HMGB1与其结合后激活TLR4/NF-κB信号通路,主要通过髓样分化因子88(myeloid differentiation factor 88,MyD88)非依赖的信号通路和MyD88依赖的信号通路[27]来参与NF-κB的活化。
2 小胶质细胞
小胶质细胞占所有胶质细胞的5%~10%,具有吞噬功能,是中枢神经系统中主要的免疫效应细胞。在正常情况下呈静息状态,形态为胞体小和细胞突起多。当受到某种信号刺激后,激活为活化型小胶质细胞,呈“阿米巴状”,其胞体呈圆形,体积增大,细胞突起缩小甚至消失,具有吞噬功能,对正常免疫应答起重要作用,吞噬堆积的碎片及消除损伤细胞[28]。但是在酸性环境改变,小胶质细胞粘附和形态变化不大,且呈现短暂下降,相对星形胶质细胞而言,继续发挥其清道夫的作用[29]。
2.1 AD中小胶质细胞的作用
小胶质细胞对神经元突触的损伤和异常积累蛋白质的免疫应答,是阿尔茨海默病的基本特征。与正常脑组织相比,AD病理显示许多激活的小胶质细胞主要聚集在斑块的内部及周围。可溶性Aβ可活化小胶质细胞,致大量的神经炎性细胞因子、炎症介质释放,包括MHC-Ⅰ类和Ⅱα、TNF-α、IL-6、IL-1、IFN-γ,α,GM-CSF、MCP-1、iNOS、NO、CD40、等[30]。通常情况下,小胶质细胞通过有效清除细胞外和早期斑块内的Aβ,以保持Aβ的产生和清除之间的平衡。然而,随着不溶性Aβ在斑块内大量聚集,显著增加小胶质细胞的活化及迁移能力,同时诱导小胶质细胞表达其炎症表面标志物CD45、CD36及CD40。在AD的后期,神经系统炎症又会反馈引起大量活化的小胶质细胞和补体的参与,而这些过多的补体及不适当激活的小胶质细胞加重了神经元细胞突触的减少,加重了AD的神经病理过程和炎症反应[31]。而其中小胶质细胞上存在大量的TLR,其在Aβ沉积导致小胶质细胞激活和清除Aβ的过程中发挥了重要作用。
2.2 TLR在AD患者脑内小胶质细胞激活中的作用
小胶质细胞上存在着大量的TLR受体,而TLR是非特异性免疫反应中识别病原体的模式识别受体,Aβ沉积与TLR结合后,导致小胶质细胞激活。也有认为TLR参与了AD小胶质细胞表型转化和调节。纤维性Aβ可以直接与TLR2、TLR4和CD14诱导作用,在AD开始阶段可以促进小胶质细胞分泌各种细胞因子、趋化因子以及对Aβ吞噬作用,但在后期增加及加速神经元凋亡[32]。无论是AD动物模型还是临床患者上,发现CD14、TLR2和TLR4的表达均会增高[33]。Richard等[34]研究发现在海马脑片内注射Aβ确实会增加TLR2基因的表达。Frank等[35]研究发现聚集淀粉样蛋白斑周围的小胶质细胞内TLR4和TLR2 mRNA含量增高。有研究认为[36],通过功能封siRNA基因沉默或者闭抗体技术抑制TLR4或TLR2,能作为治疗靶点,能够有效阻止小胶质细胞的激活,TNF-α和IL-6的产生。神经干细胞移植后,APP/PS1转基因小鼠明显改善认知障碍,并通过抑制小胶质细胞上TLR4介导的炎症通路激活[37]。有研究表明[38]经过Aβ预处理的野生型小胶质细胞释放的炎性介质具有明显神经毒性,而预处理的CD-14或者TLR-4缺失的小胶质细胞不再有神经毒性,证明TLR参与了Aβ激活小胶质细胞释放神经毒性介质的过程。
2.3 TLR在小胶质细胞清除Aβ中的作用
TLR和CD14相互作用共同参与了小胶质细胞吞噬Aβ蛋白的过程。TLR2和TLR4也是纤维化Aβ激活小胶质细胞吞噬作用所必需的[39]。虽然在TLR与Aβ结合后,早期小胶质细胞能够有效吞噬AD形成的Aβ和破损的神经元;但在AD晚期,由于Aβ的不断累积导致了小胶质细胞大量释放炎症介质和Aβ清除机制的基因下调[40]。
3 AD中HMGB1与小胶质细胞的关系
小胶质细胞在AD的病程进展中起到了“双刃剑”的作用。在AD发病早期,小胶质细胞对神经起到保护作用,能够清除Aβ。RAGE、TLR2、TLR4在AD小胶质细胞的激活及Aβ清除过程中发挥着重要作用[41,42]。与此同时,小胶质细胞产生许多促炎因子和免疫介质从而为AD的病程进展创造了一个神经毒性的环境[43,44]。最近的研究[45]指出HMGB1是造成神经退行性疾病的一项危险因素。在AD中,细胞外的HMGB1通过抑制小胶质细胞的吞噬作用和稳定Aβ低聚体从而有助于AD的病程进展。在P35敲除的小鼠上,在海马区可以发现Aβ沉积的区域能引起大量的小胶质细胞的激活,且小胶质细胞的浸润可能导致可溶性高迁移率族蛋白B1(HMGB1)分泌增加[46]。Takata等[8]发现在AD模型中,大鼠小胶质细胞能吞噬Aβ42以及HMGB1抑制吞噬作用。是通过HMGB l通过紧密结合Aβ42,使Aβ42处于稳定的寡聚体状态,减弱小胶质细胞对Aβ42清除的能力。而在其随后研究中发现,给予外源性的HMGB1使小胶质细胞胞质中Aβ40增加后,其延迟期降解,导致其胞外的Aβ沉积,表明在积累Aβ斑块细胞外HMGB1引起的小胶质细胞吞噬功能异常是AD重要形成原因之一[20]。Mazarati等[45]发现在AD神经系统内,高水平状态的HMGB l可介导的炎症反应导致神经变性疾病的发生。且HMGB1是通过RAGE和TLRs介导的[47]产生的神经炎症反应。
在AD早期可出现HMGB1含量增加,与其受体(RAGE和TLR)结合后,可识别及跨膜信号转导,介导细胞内NF-κB的通路,引起大量炎症介质释放,并抑制小胶质细胞对Aβ的清除和增强Aβ的神经毒性。而小胶质细胞可通过其TLR受体激活,吞噬过多沉积Aβ。两者之间失衡也是AD发生及进展的重要原因。因此HMGB1可能成为治疗AD的一种新的靶标。
[1]Crews L,Masliah E.Molecular mechanisms of neurodegeneration in Alzheimer's disease[J].Hum Mol Genet,2010,19(R1):R12-20.
[2]Selko DJ.Translating cell biology into therapeutic advances in Alzheimer's disease[J].Nature,1999,399(6738 Suppl):A23-31.
[3]Akiyama H,Barger S,Barnum S,et al.Inflammation and Alzheimer's disease[J].Neurobiol Aging,2000,21:383-421.
[4]Nicoll JA,Wilkinson D,Holmes C,et al.Neuropathology of human Alzheimer disease after immunization with amyloid-peptide:A case report[J].Nat Med,2003,9(4):448-452.
[5]Lee L,Kosuri P,Arancio O.Picomolar amyloid-β peptides enhance spontaneous astrocyte calcium transients[J].J Alzheimers Dis,2014,38(1):49-62.
[6]Akiyama H,McGeer PL.Specificity of mechanism for plaque removal after aimmunotherapy for Alzheimer disease[J]. Nat Med,2004,10(2):118-119.
[7]Minett T,Classey J,Matthews FE,et al.Microglial immunophenotype in dementia with Alzheimer's pathology[J]. J Neuroinflammation,2016,13(1):135.
[8]Takata K,Kitamura Y,Kakimura J,et al.Role of high mobility group protein-1(HMG1)in amyloid-beta homeosta sis[J].Biochem Biophys Res Commun,2003,301(3):699-703.
[9]Takata K,Kitamura Y,Tsuchiya D,et al.High mobility group box protein-1 inhibits microglial Abeta clearance and enhances Abeta neurotoxicity[J].J Neurosci Res,2004,78(6):880-891.
[10]Rauvala H,Rouhiainen A.RAGE as a receptor of HMGB1(Amphoterin):Rolesinhealthanddisease[J].CurrMolMed,2007,7(8):725-734.
[11]刘宇,曹清心,赵仙先,等.高迁移率族蛋白B1与心血管疾病相关性的研究进展[J].医学研究生学报,2012,25(1):99-102.
[12]Wang FC,Pei JX,Zhu J,et al.Overexpression of HMGB1 A-box reduced lipopolysaccharide-induced intestinal inflammation via HMGB1/TLR4 signaling in vitro[J].World J Gastroenterol,2015,21(25):7764-7776.
[13]Muth IE,Zschüntzsch J,Kleinschnitz K,et al.HMGB1 and RAGE in skeletal muscle inflammation:Implications for protein accumulation in inclusion body myositis[J]. Exp Neurol,2015,271:189-197
[14]Zeng JC,Xiang WY,Lin DZ,et al.Elevated HMGB1-related interleukin-6 is associated with dynamic responses of monocytes in patients with active pulmonary tuberculosis[J].Int J Clin Exp Pathol,2015,8(2):1341-1353.
[15]刘旺华,李花,肖献忠,等.高迁移率族蛋白B-1与炎症关系的研究进展[J].中国病理生理杂志,2008,24(8):1656-1660.
[16]Vogel S,Bodenstein R,Chen Q,et al.Platelet-derived HMGB1 is a critical mediator of thrombosis[J].J Clin Invest,2015,125(12):4638-4654.
[17]Tang D,Kang R,Zeh HJ 3rd,et al.High-mobility group box 1,oxidative stress,and disease[J].Antioxid Redox Sign,2011,14(7):1315-1335.
[18]Yang X,Wang H,Zhang M,et al.HMGB1:A novel protein that induced platelets active and aggregation via Toll-like receptor-4,NF-κB and cGMP dependent mechanisms[J].Diagn Pathol,2015,10:134.
[19]戴丽,杨芝春,李元建,等.糖基化终产物与糖尿病血管并发症[J].国际病理科学与临床杂志,2012,32(1):30-34.
[20]Takata K,Takada T,Ito A,et al.Microglial amyloid-β 1-40 phagocytosis dysfunction is caused by high-mobility group box protein-1:Implications for the pathological progression of Alzheimer's disease[J].Int J Alzh-eimers Dis,2012,2012:685739.
[21]Meneghini V,Bortolotto V,Francese MT,et al.High-mobility group box-1 protein and β-amyloid oligomers promote neuronal differentiation of adult hippocampal neural progenitors via receptor for advanced glycation end products/nuclear factor-κB axis:Relevance for Alzh-eimer's disease[J].J Neurosci,2013,33(14):6047-6059.
[22]Bonini SA,Ferrari-Toninelli G,Uberti D,et al.Nuclear factor κB-dependent neurite remodeling is mediated by notch pathway[J].J Neurosci,2011,31(32):11697-11705.
[23]Koo JW,Russo SJ,Ferguson D,et al.Nuclear factorkappa B is a critical mediator of stress-impaired neuroge nesis and depressive behavior[J].Proc Natl Acad Sci US A,2010,107(6):2669-2674.
[24]张婷,夏敏.高迁移率族蛋白B1信号转导通路的研究进展[J].医学综述,2011,17(2):195-198.
[25]Van Beijnum JR,Buurman WA,Griffioen AW,et al. Convergence and amplification of toll-like receptor(TLR)and receptor for advanced glycation end products(RAGE)signaling pathways via high mobility group B1,HMGB1)[J].Angiogenesis,2008,11(1):91-99.
[26]Trotta T,Porro C,Calvello R,et al.Biological role of tolllike receptor-4 in the brain[J].J Neuroimmunol,2014,268(1-2):1-12
[27]Akira S,Takeda K.Toll-like receptor signalling[J].NatRev Immunol,2004,4(7):499-511.
[28]Bruce Keller AJ.Microglial-neuronal interactions in synaptie damage and recovery[J].J Neurosci Res,1999,58(1):191-201.
[29]Eugenín J,Vecchiola A,Murgas P,et al.Expression pattern of scavenger receptors and amyloid-β phagocytosis of astrocytes and microglia in culture are modified by acidosis:Implications for Alzheimer's disease[J].J Alzheimers Dis,2016,52(3):1-17.
[30]Browne TC,McQuillan K,McManus RM,et al.IFN-γ production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer's disease[J].J Immunol,2013,190(5):2241-2251.
[31]Hong S,Beja-Glasser VF,Nfonoyim BM,et al.Complementandmicrogliamediateearlysynapselossin Alzheimer mouse models[J].Science,2016,352(6286):712-716.
[32]Gambuzza ME,Sofo V,Salmeri FM,et al.Toll-like receptors in Alzheimer's disease:A therapeutic perspective[J]. CNS Neurol Disord Drug Targets,2014,13(9):1542-1558.
[33]Zhang W,Wang LZ,Yu JT,et al.Increased expressions of TLR2 and TLR4 on peripheral blood mononuclear cells from patients with Alzheimer's disease[J].J Neurol Sci,2012,315(1-2):67-71.
[34]Richard KL,Filali M,Préfontaine P,et al.Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta1-42 and delay the cognitive decline in a mouse model of Alzheimer's disease[J].J Neurosci,2008,28(22):5784-5793.
[35]Frank S,Copanaki E,Burbach GJ,et al.Differential regulation of toll-like receptor mRNAs in amyloid plaqueassociated brain tissue of aged APP23 transgenic mice[J]. Neurosci Lett,2009,453(1):41-44.
[36]Udan ML,Ajit D,Crouse NR,et al.Toll-like receptors 2 and 4 mediate Abeta(1-42)activation of the innate im mune response in a human monocytic cell line[J].J Neurochem,2008,104(2):524-533.
[37]Zhang Q,Wu HH,Wang Y,et al.Neural stem cell transplantation decreases neuroinflammation in a transgenic mouse model of Alzheimer'sdisease[J].J Neurochem,2016,136(4):815-825.
[38]Mandrekar-Colucci S,Landreth GE.Microglia and inflammation in Alzheimer's disease[J].CNS Neurol Disord DR,2010,9(2):156-167.
[39]Reed-Geaghan EG,Savage JC,Hise AG,et al.CD14 and toll-like receptors 2 and 4 are required for fibrillar Aβstimulated microglial activation[J].J Neurosci,2009,29(38):11982-11992.
[40]Hickman SE,Allison EK,EI Khoury J,et al.Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice[J].J Neurosci,2008,28(33):8354-8360.
[41]Lue LF,Walker DG,Brachova L,et al.Involvement of microglial receptor for advanced glycation endproducts,RAGE)in Alzheimer's disease:Identification of a cellu lar activation mechanism[J].Exp Neurol,2001,171(1):29-45.
[42]Tahara K,Kim HD,Jin JJ,et al.Role of toll-like receptor signalling in Abeta uptake and clearance[J].Brain,2006,129(11):3006-3019.
[43]Akiyama H,Barger S,Barnum S,et al.Inflammation and Alzheimer's disease[J].Neurobiol Aging,2000,21(3):383-421.
[44]Wyss-Coray T.Inflammation in Alzheimer disease:Driving force,bystander or beneficial response[J].Nat Med,2006,12(9):1005-1015.
[45]Mazarati A,Maroso M,Iori V,et al.High-mobility group box-1 impairs memory in mice through both toll like receptor 4 and receptor for advanced glycation end products[J].Exp Neurol,2011,232(2):143-148.
[46]Jang A,Liew H,Kim Y,et al.P35 deficiency accelerates HMGB-1-mediated neuronal death in the early stages of an Alzheimer's disease mouse model[J].MCurr Alzheimer Res,2013,10(8):829-843.
[47]Rauvala H,Rouhiainen A.Physiological and pathophysiological outcomes of the interactions of HMGB1 with cell surface receptors[J].Biochim Biophys Acta,2010,1799(1-2):164-170.
HMGB1 and microglia in the role of in the development and progression of Alzheimer's disease
KONG WeiweiLIU XuhuaCAO HongLI Jun
Department of Anesthsiology,the Second Affiliated Hospital of Wenzhou Medical University,Wenzhou 325027,China

图1 原发性肝癌动脉期(见内文第28页)
图2 原发性肝癌门静脉期(见内文第28页)

图3 原发性肝癌实质期(见内文第28页)

图4 TGF-β1在小鼠肺纤维化中的表达(SP×400)(见内文第32页)

图5 α-SMA在小鼠肺纤维化中的表达(SP×400)(见内文第32页)

图6 32例儿童及青少年鼻咽癌生存曲线(见内文第71页)
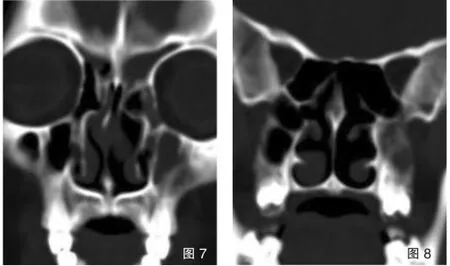
图7、8 为同一上颌窦内两种分隔。同一病例不同层面冠状位图像,分别显示出右侧上颌窦内存在两种分隔,即垂直矢状分隔(图7)及水平分隔(图8);同时显示对侧上颌窦及双侧筛窦内炎症改变(见内文第101页)
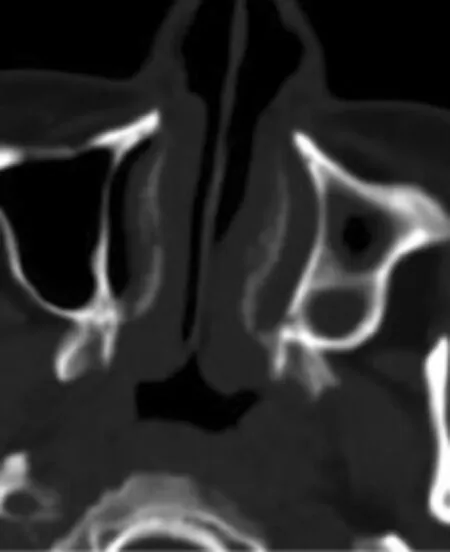
图9 垂直冠状分隔横断面图像显示左侧上颌窦的冠状分隔(见内文第101页)


图10、11 为垂直矢状分隔。同一病例,左侧上颌窦垂直矢状分隔在横断面显示为鼻窦前壁骨嵴样突起(图10白箭),多平面重组(MPR)冠状位显示出窦内分隔全貌(图11)(见内文第101页)

图12、13 为水平分隔。图12显示双侧上颌窦内的水平分隔,可见双侧上颌窦下方的分隔腔开口于中鼻道;同时可见双侧上颌窦内的炎症改变;图13另一例病例显示双侧上颌窦内的水平分隔,可见双侧上颌窦上方的分隔腔开口于中鼻道;同时可见双侧上颌窦内各分隔腔内的炎症改变(见内文第101页)
图14 斜行分隔横断位显示左侧上颌窦内的斜行分隔(见内文第101页)
R749.16
A
1673-9701(2016)22-0164-05
2016-06-13)
国家自然科学基金(81271204);浙江省公益技术应用研究计划项目(2016C37098)
