X-连锁遗传性耳聋*
2016-10-19王秋菊王洪阳
王秋菊 王洪阳
X-连锁遗传性耳聋是指与耳聋表型相关的致病基因位于X染色体上,这些基因的遗传方式可以是显性的,也可以是隐性的,此类耳聋表型与性别相关。1930年Dow和Poyner首次报道了典型的X-连锁遗传性耳聋,随后发现与常染色体遗传性耳聋相同,X-连锁遗传性耳聋也包括非综合征型耳聋及综合征型耳聋。对于非综合征型X-连锁遗传性耳聋,听力损失是其唯一临床表现,根据显性及隐性遗传方式的不同,患者的发病年龄、听力损失程度等表型亦各异。综合征型X-连锁遗传性耳聋,除了耳聋表型外同时伴有其他器官系统功能异常,比如Alport综合征、Norrie综合征和伴外淋巴井喷的进行性混合性聋都是X-连锁遗传病。X-连锁遗传性耳聋较常染色体遗传性耳聋相对罕见,在遗传性耳聋中的发病比例在1%左右,但在临床遗传咨询中,通过家系图谱分析、患者的表型特征及基因检测明确X-连锁遗传,尤其是要明确家系中女性患者和女性携带者,这对减少家庭中后代耳聋的发生意义重大。
·继续教育园地·
X-连锁遗传性耳聋*
王秋菊1王洪阳1
X-连锁遗传性耳聋是指与耳聋表型相关的致病基因位于X染色体上,这些基因的遗传方式可以是显性的,也可以是隐性的,此类耳聋表型与性别相关。1930年Dow和Poyner首次报道了典型的X-连锁遗传性耳聋,随后发现与常染色体遗传性耳聋相同,X-连锁遗传性耳聋也包括非综合征型耳聋及综合征型耳聋。对于非综合征型X-连锁遗传性耳聋,听力损失是其唯一临床表现,根据显性及隐性遗传方式的不同,患者的发病年龄、听力损失程度等表型亦各异。综合征型X-连锁遗传性耳聋,除了耳聋表型外同时伴有其他器官系统功能异常,比如Alport综合征、Norrie综合征和伴外淋巴井喷的进行性混合性聋都是X-连锁遗传病。X-连锁遗传性耳聋较常染色体遗传性耳聋相对罕见,在遗传性耳聋中的发病比例在1%左右,但在临床遗传咨询中,通过家系图谱分析、患者的表型特征及基因检测明确X-连锁遗传,尤其是要明确家系中女性患者和女性携带者,这对减少家庭中后代耳聋的发生意义重大。
1 X-连锁遗传方式的主要特征
①交叉遗传:男性患者的X-连锁基因只能来源于母亲并只能传给女儿;
②半合子:正常男性只有一条X染色体,相当于正常女性的一半,因此称为“半合子”,位于男性X染色体上的致病基因突变,无论显性隐性,均能导致疾病的发生;
③女性表型差异:X-连锁遗传基因在女性中是否表达及表达程度与X染色体失活相关,X染色体失活具有选择性和偏好性。
1.1X-连锁隐性遗传(X-linked recessive inheritance)的特点是:①男性患者远多于女性患者;②男性患者的双亲都无病,其致病基因来自携带者母亲;③可见交叉遗传现象,即“父传女,母传子”的遗传现象;④由于男患者的子女都是正常的,所以代与代之间可见明显的不连续现象,即隔代遗传。1995年鉴定的第一个非综合征型耳聋基因POU3F4相关耳聋基因遗传方式即表现为X-连锁隐性遗传。
1.2X-连锁显性遗传(X-linked Dominant Inheritance)的特点是:①女性患者多于男性患者;②患者双亲之一必定是患者,女患者都是杂合子,她们的致病基因可传给儿子和女儿,但男患者的致病基因只传给女儿,因此,系谱中男患者的女儿全部发病;③与常染色体显性遗传相似,家系中可看到连续两代以上都有患者。
2 X-连锁遗传性耳聋致病基因的命名与定位
迄今为止, X-连锁遗传性耳聋中只有6个相关基因座被命名与定位。根据人类基因组命名委员会的规则,以DFNX为前缀来表示X-连锁遗传性耳聋,DFN缩略词来自deafness(耳聋)中的三个字母,X定义为X-连锁遗传。第一个致病基因座位表述为DFNX1,此后依据基因座发现及获得命名的时间顺序排列,6个X-连锁遗传的非综合征型耳聋相关基因座依次命名为DFNX1、DFNX2、DFNX3、DFNX4、DFNX5、DFNX6。
X-连锁基因座位命名早期以DFN为前缀,分别命名为DFN1, DFN2,DFN3,DFN4,DFN5,DFN6,DFN7,DFN8,现今的基因座位命名与原始命名的关系详见表1。除DFN2, 3, 4, 6有新的命名外,DFN1重新定义为综合征表型,该基因座位最早定位于1960年,Mohr和Mageroy报道了一个挪威家系,表现为X-连锁的进行性下降的感音神经性耳聋,男性患者在儿童期就表现出明显听力障碍和言语困难并逐渐加重,女性携带者则偶尔出现轻度听力障碍,通过连锁分析定位于X染色体,命名为DFN1;1995年,Tranebjaerg对该家系表型进行追踪分析,发现该家系实际是一种隐性神经退化综合征,其表型特征为在10岁前以渐进下降的语后感音神经性耳聋为首发症状,随后出现肌张力障碍、痉挛状态、吞咽困难、智力迟滞、偏执症及皮层盲,该综合征的致病基因为TMM8A(MIM300356)。因此,目前仍以新的命名方式DFNX确定X连锁遗传性耳聋的表型和致病基因。
3 非综合征型X-连锁线性耳聋相关致病基因的鉴定及表型(表1)
3.1DFNX1(原DFN2)与PRPS1基因
DFNX1定位在Xq22,表型以重度先天性感音神经性耳聋为主,致病基因为PRPS1基因。遗传方式为X-连锁显性遗传,男性患者表型较重,女性携带者部分可表现为轻度到中度听力损失。1996年,Tyson等在一个四代遗传的英国家系中定位了DFN2,家系内所有男性患者表现为先天性双侧重度到极重度感音神经性耳聋,女性携带者无听力障碍主诉,但听力学检测显示高频的轻度到中度听力下降。随后报道的DFN2家系表现为语后渐进性下降的非综合征型感音神经性耳聋。2010年,袁慧军等在一个中国家系的男性患者中鉴定了致病基因PRPS1,随后在中国、美国、英美家庭中分别鉴定了该基因与非综合征型X-连锁遗传性耳聋相关的致病突变。
PRPS1基因编码磷酸核糖焦磷酸合成酶,催化5-磷酸核糖和ATP的反应,生成AMP和PRPP(5-磷酸核糖-1-焦磷酸),对于嘌呤、嘧啶和合成吡啶核苷酸的生成和重新利用过程是必不可少的。PRPS1基因突变会导致酶活性降低,小鼠实验中,Prps1在小鼠耳蜗、前庭毛细胞及螺旋神经节表达,且在毛细胞中持续表达,出生后在螺旋神经节中仍有表达。除与非综合征型耳聋相关外,PRPS1亦为综合征型耳聋CMTX5的致病基因,除耳聋外,伴随有视力下降和周围神经病变(表2)。
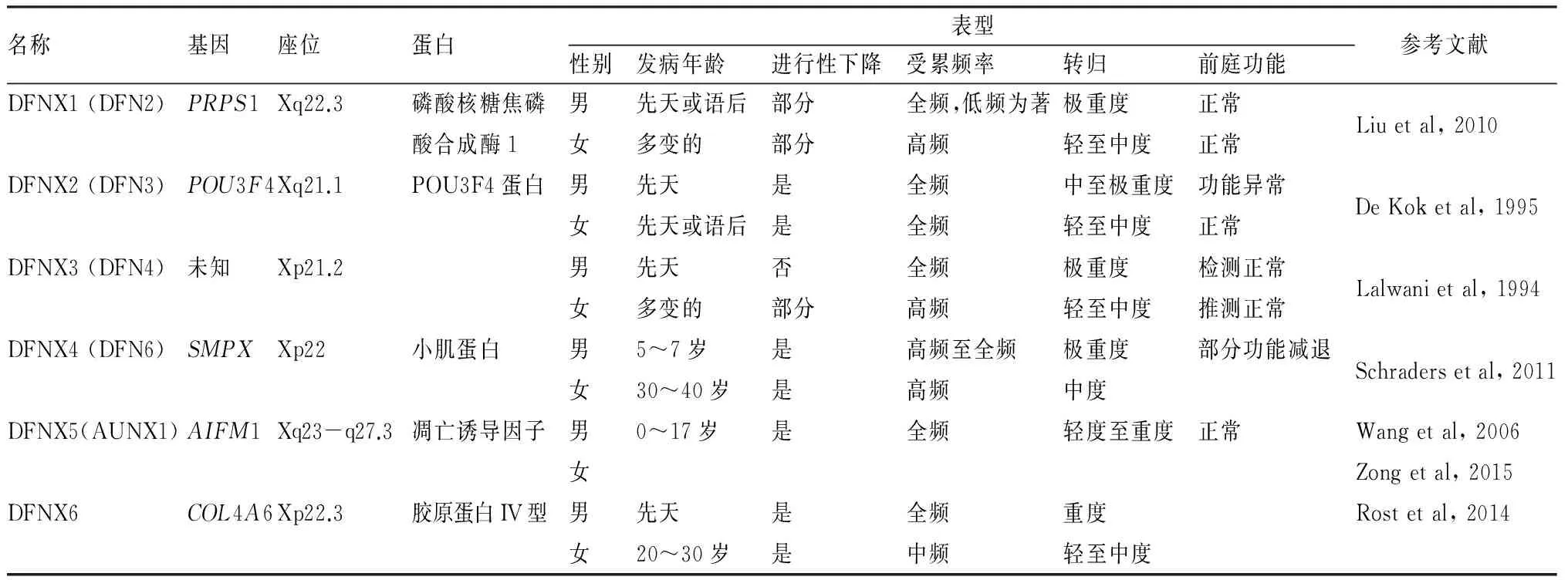
表1 非综合征型X-连锁性遗传性耳聋的相关基因、定位、表达及表型

表2 综合征型X-连锁性遗传性的相关基因、定位、表达及表型
3.2DFNX2(原DFN3)与POU3F4基因
DFNX2定位在Xq13-q21,表型为感音神经性聋或混合性聋及镫骨手术时的外淋巴井喷,致病基因为POU3F4基因。遗传方式为X-连锁隐性遗传,患者以男性为主,表现为中度到极重度感音神经性听力损失,或混合性听力损失伴感音神经性成分的进行性发展,部分女性携带者中亦可见轻度的感音神经性或混合性听力损失,且可能呈进行性发展,影像学可见耳蜗基底回和内听道之间的骨壁变薄,内听道侧壁末端扩大。
POU3F4基因属于POU转录因子家族成员中的第三类,POU转录因子对于器官形成、细胞分化发挥着重要作用。Minowa等在DFN3老鼠模型中发现老鼠中耳发育正常,但是听性脑干反应阈值增加;进一步研究发现其内耳螺旋韧带中的纤维细胞发生形态学改变,而内耳中其他组织细胞没有发生变化,故认为Brn4基因(人类POU3F4基因的同源基因)突变导致纤维细胞功能异常,而纤维细胞在维持耳蜗内电位中发挥重要的作用。Phippard等研究也证明Brn4基因产物是内耳中胚层间充质的塑型关键因子,Brn4基因突变会导致耳蜗、上半规管等结构发育异常。
3.3DFNX3(原DFN4)
DFNX3定位在Xp21.2,在此区域包括杜氏肌营养不良的座位,这个座位突变在男性可以导致先天性极重度感音神经性聋,在女性可以导致成年发病的轻度到中度的高频感音神经性聋,是已经定位的6个X-连锁基因座位中唯一尚未克隆致病基因的座位。
3.4DFNX4(原DFN6)与SMPX基因
DFNX4定位在Xp22,表型以进行性感音神经性聋为主,致病基因为SMPX,遗传方式为X-连锁显性遗传。2011年,Antje等报道了一个德国非综合征型感音神经性听力损失大家系,临床表现为男性3~7岁发病,女性20~30余岁发病,由高频轻度听力损失逐渐发展为全频重度听力损失。对这个家系进行分子遗传学研究,在患者中发现了位于SMPX基因第3外显子上的一个新的无义突变c.109G>T(p.Glu37X),与耳聋表型共分离。同时,在一个具有相似表型的西班牙家系中发现了SMPX基因第4外显子的一个无义突变c.175G>T(p.Gly59X)。同年,Margit等在X-连锁语后进行性听力损失的一个荷兰五代大家系,临床表现为双侧进行性听力损失,运用分子遗传学方法,在SMPX的外显子4上,发现了1个c.214G>T无义突变(p.Glu72X),这三个无义突变均导致了蛋白编码提前终止。
SMPX基因编码小肌肉蛋白(small muscular protein),目前尚未明确其功能结构域。1999年,Dirk Patzak等在人类心脏和骨骼肌发现SMPX基因的特异性表达,同时,在小鼠模型的研究中发现其除了在心脏和骨骼肌大量表达外,在肝,睾丸,肾脏,脑等组织内也有少量分布。 2010年,Heejei Yoon等在胚胎小鼠内耳组织发现Smpx基因在内耳毛细胞明确表达,并随发育时间的变化其表达显著改变,Smpx最先在耳蜗半规管的底回和中间回高度表达,而在中间-顶回表达微弱,出生后第5天,Smpx在耳蜗管各回均强烈表达。这些改变提示SMPX基因不仅是肌细胞运动功能的重要基因,而且提示其在毛细胞分化和功能中也起到重要的作用。SMPX基因编码的小肌肉蛋白可能与耳蜗毛细胞纤毛的发育密切相关。这些微细结构会对声波产生反应,并将声音转变成电能输送到大脑的听觉中枢,因此,毛细胞上这些静纤毛出现问题会引起听力损害。内耳细胞长期接受机械性压力,而其正常状态和功能的维护有赖于SMPX。
3.5DFNX5(原AUNX1)与AIFM1基因
DFNX5定位在Xq23-q27.3,患者表现为听神经病表型,听力损失以轻度至重度感音神经性耳聋为主,部分患者伴有其他周围神经病变;遗传方式为X-连锁隐性遗传,患者以男性为主。2003年,王秋菊等发现一个交叉遗传男性发病的听神经病伴迟发性、周围神经病大家系,在国际上首次描述了遗传性听神经病的X-连锁隐性遗传方式及中国大家系的表型特征;2004年,通过连锁分析的方法,将该家系表型定位在X染色体上Xq23-q27.3区域,将其命名为AUNX1基因座;2011年,利用最新的全外显子组二代测序技术,结合基因定位信息,成功鉴定出了与家系表型共分离的致病基因(AUNX1)及其错义突变p.R451Q。
AIFM1基因编码蛋白与线粒体氧呼吸链功能密切相关,功能预测发现,这些新突变将明显改变编码蛋白的表面构象及结合力,从而严重影响蛋白的生理功能。该基因在内耳毛细胞、螺旋神经节上均有表达,提示其在听觉系统中可能发挥重要作用。
3.6DFNX6与COL4A6基因
DFNX6定位在Xq22.3,表型以重度感音神经性聋为主,致病基因为COL4A6,遗传方式为X-连锁隐性遗传,患者以男性为主,女性携带者不出现听力下降或表现为轻度至中度听力下降。2014年,Rost等在一个匈牙利家系中鉴定了COL4A6为该家系的致病原因,家系成员除耳聋表型外,无血管球性肾炎、肾衰、视觉受损等其他系统受损表现。
COL4A6基因编码基膜的IV型胶原的α6链,与COL4A5编码的两条α5链形成异源三聚体。COL4A5基因突变及X染色体上包含COL4A5及COL4A6基因的片段缺失与Alport综合征相关(表2)。在小鼠中主要表达在螺旋韧带的血管纹处,斑马鱼胚胎中Col4a6在神经系统及耳部尤其是耳泡部位动态表达,维持正常耳朵的发育与功能。
4 综合征型X-连锁遗传性耳聋相关基因、定位及表达
表2列举了部分综合征型X-连锁耳聋相关基因、定位及表达。以X-连锁进行性神经性肌萎缩伴耳聋(CMT综合征)为例,1993年, Bergoffen等在CMTX1家系中鉴定GJB1为致病基因。GJB1基因是与X-连锁遗传性感觉运动性神经病伴耳聋相关的致病基因。人类的GJB1基因位于染色体Xq13.1区域,DNA序列全长1 969 bp,只有1个外显子,编码长度为283 aa的缝隙连接蛋白Cx32,该蛋白有4个跨膜区、2个胞外突起和3个胞内片段。LoPez-Bigas等研究发现,Cx32在发育中的鼠听泡上皮仅在有限的区域表达,但是在周围间叶组织却有大量的转录,随着耳蜗结构的不断发育完善,Cx32逐渐分布于螺旋韧带、前庭膜、基底膜和螺旋神经节边缘等处。Forge等发现Cx32在前庭器支持细胞、膝神经节和听神经也有表达。研究显示,Cx32可以在髓鞘形成通道, 运输一些离子、营养成分等小分子进入神经细胞, 在营养神经及信号传导中均可起到重要作用, 其在中枢神经系统、周围神经的雪旺氏细胞均有表达。GJB1基因敲除小鼠表现力一种轻微的、迟发的外周神经病变,其特征为髓鞘变薄、洋葱球样细胞形成和突触旁囊泡扩大。
5 X-连锁遗传性耳聋的婚配类型及子代发病风险的预测与遗传咨询
X-连锁隐性遗传耳聋患者,如父亲正常(XAY),母亲为携带者(XAXa),即能产生两种配子A和a,一种带有正常基因A,一种带有突变基因a,后代儿子发病风险为50%(XAY与XaY两种可能),表型正常的女性为致病基因突变的携带者风险为50%(XAXa与XAXA两种可能);如果父亲患病(XaY),母亲正常,如果母亲不携带致病突变(XAXA),则后代儿子无患病风险,而女儿全部为携带者,如果母亲携带致病突变(XAXa),后代儿子发病风险为50%,女儿为携带者及发病的风险分别为50%。在发现X-连锁隐性遗传耳聋家系时,明确家系中的女性是否为致病基因的携带者至关重要,可以由此判断出后代的发病情况。
X-连锁显性遗传的父母至少一方为患者,如果父亲为患者(XAY),母亲正常(XaXa)时,则其所有女性后代均患病,所有男性后代均正常;如果母亲为患者,父亲正常,母亲为杂合子(XAXa)时,其子女各有50%的患病风险,母亲为纯合子(XAXA)时,后代中儿女全部患病。
1Toriello HV, Reardon W, Gorlin RJ. Hereditary hearing loss and its syndromes (Second edition)[M]. The United States of America:Oxford University Press, 2004.1~2.
2Cryns K, Van Camp G. Deafness genes and their diagnostic applications[J]. Audiol Neurootol,2004,9:2.
3http://hereditaryhearingloss.org/.
4Liu X, Han D, Li J, et al. Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2[J]. Am J Hum Genet, 2010,86:65.
5Wang QJ, Li QZ, Rao SQ, et al. A Novel Mutation of POU3F4 Causes Congenital Profound Sensorineural Hearing Loss in a Large Chinese Family[J]. Laryngoscope,2006,116:944.
6Wang QJ, Li QZ, Rao SQ, et al. AUNX1, a novel locus responsible for X-linked recessive auditory and peripheral neuropathy, maps to Xq23-27.3[J]. J Med Genet,2006, 43:1.
7Schraders M, Haas SA, Weegerink NJD, et al. Next-generation sequencing identifies mutations of SMPX, which encodes the small muscle protein, X-linked, as a cause of progressive hearing impairment[J]. Am J.Hum Genet, 2011, 88:628.
8Zong L, Guan J, Ealy M, et al. Mutations in apoptosis-inducing factor cause X-linked recessive auditory neuropathy spectrum disorder[J]. J Med Genet, 2015, 52:523.
9Rost S, Bach E, Neuner C, et al. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6[J]. Europ J Hum Genet,2014,22:208.
(2016-08-19收稿)
(本文编辑周涛)
*国家重大科学研究计划项目(2014CB943001)、国家自然基金重点国际合作项目(81120108009)、国家自然科学基金重点项目(81530032)联合资助
1解放军总医院耳鼻咽喉头颈外科 解放军耳鼻咽喉研究所 (北京100853)
王秋菊(Email:wqcr@263.net 或wqcr301@vip.sina.com)
10.3969/j.issn.1006-7299.2016.05.021
R764.43
A
1006-7299(2016)05-0520-04
