在CPS微球表面固载氨基酚型双齿席夫碱氧钒(Ⅳ)配合物及其催化氧化性能
2016-10-17孟素青崔坤俐高保娇
孟素青, 崔坤俐, 高保娇
(中北大学化工系,太原 030051)
在CPS微球表面固载氨基酚型双齿席夫碱氧钒(Ⅳ)配合物及其催化氧化性能
孟素青,崔坤俐,高保娇
(中北大学化工系,太原 030051)
使乙醛酸(GA)与氯甲基化交联聚苯乙烯(CMCPS)微球发生酯化反应,将醛基(AG)引入交联聚苯乙烯(CPS)微球表面,得到改性微球CPS-AG;再以间氨基苯酚(MAP)为试剂,使微球CPS-AG表面的AG发生席夫碱反应,制得了表面键合有氨基酚型双齿席夫碱配基的功能微球CPS-AGAP;最后,使微球CPS-AGAP与硫酸氧钒发生配位螯合反应,获得了表面固载有氨基酚型双齿席夫碱氧钒(Ⅳ)配合物的固体催化剂微球CPS-[VO(AGAP)2]。重点考察了主要因素对GA与CMCPS微球的酯化反应的影响。采用红外光谱(FT-IR)、固体紫外(UV)及扫描电子显微镜(SEM)对催化剂微球进行了充分表征。分别将微球CPS-[VO(AGAP)2]用于环己醇和乙苯的分子氧氧化过程,考察其催化活性。实验结果表明,溶剂的极性有利于GA与CMCPS微球之间的酯化反应,极性较强的N,N-二甲基乙酰胺为适宜的反应溶剂,90 ℃为适宜的反应温度。在适宜的反应条件下,CMCPS微球的氯甲基转化率可以达到82%。在分子氧氧化环己醇和乙苯的过程中,非均相催化剂CPS-[VO(AGAP)2]微球均表现出良好的催化活性。
交联聚苯乙烯微球; 双齿席夫碱配基; 氧钒(Ⅳ)配合物; 固载; 催化氧化
以分子氧为氧化剂,采用有效的催化剂,可以实现各种类型有机物的氧化转变,比如将烷烃与烯烃转变为含氧化合物[1-2],将醇类物质转变为羰基化合物[3-4]以及使含硫化合物进行的氧化转变等[5-6],此过程环境友好,且具有原子经济性的特点[7-8],在有机合成研究及工业化生产中都备受关注。在各种类型的分子氧氧化催化剂中,过渡金属配合物催化剂占有重要的位置,仿生催化剂金属卟啉也属于此类催化剂。在多种过渡金属元素中,高价态(Ⅴ、IV)的氧钒配合物具有很强的催化氧化活性[9-10],各种配基的氧钒配合物催化剂被广泛研究与开发,其中,由于以席夫碱(Schiff base)化合物为配基的过渡金属配合物在结构和催化氧化活性上与细胞色素P-450中的金属卟啉类似,故近年来,席夫碱氧钒配合物催化剂的研究十分活跃,广泛应用于各类有机物的分子氧催化氧化过程中[11-13]。席夫碱化合物中可含有2个或多个配原子,从而可形成双齿(N,O型、N,N型、N,S型等)或多齿席夫碱金属配合螯合物,以这样的配基所形成的配合物具有更稳定的物理化学性能。
目前,研究与应用的席夫碱氧钒配合物大多为均相催化剂,存在催化剂难以分离回收与循环使用的缺点,而且也影响产物的分离与纯化,引起环境污染,故导致催化氧化的总效率比较低。变均相催化剂为非均相催化剂,即实现均相催化剂的固载化可有效克服上述缺点,使化学合成过程绿色化[14-15]。已有研究者分别以沸石、蒙脱土、聚合物树脂等为载体,制备了非均相的席夫碱氧钒配合物催化剂[16-18],但文献报道的尚且很少。交联聚苯乙烯(CPS)微球由于具有价格低廉、力学性能与化学稳定性良好,易于进行化学修饰[19-20]等优点而被广泛用作催化剂载体。本课题组在前期的研究中,通过大分子反应途径,研究制备了CPS微球固载的水杨醛型(N,O型)[21]、氨基吡啶型(N,N型)双齿席夫碱氧钒配合物固体催化剂[22]。
本文采用独特的大分子反应途径,先使乙醛酸(GA)与氯甲基化交联聚苯乙烯(CMCPS)微球发生酯化反应,从而将醛基(AG)引入微球表面;然后以氨基苯酚(AP)为试剂,通过席夫碱反应制得了键合有氨基苯酚型双齿席碱配基的功能微球CPS-AGAP;最后使其与硫酸氧钒发生配位螯合反应,制备了固载有席夫碱氧钒(Ⅳ)配合物的固体催化剂CPS-[VO(AGAP)2]。与文献[22]中固体催化剂(N,N型)的制备相比,本文制备的产物属N,O型席夫碱氧钒配合物固体催化剂,其大分子反应途径不同,且使用的主要化学试剂氨基苯酚价格低廉,制备途径独特,催化剂成本低;与文献[21]中固体催化剂的制备相比,虽然固载的席夫碱氧钒配合物均属N,O型,但本文制备固体催化剂的路线更为简捷,类似的研究尚鲜见文献报道。本文重点研究在CPS微球表面键合氨基苯酚型双齿席夫碱氧钒配合物的大分子反应过程,并分别以环己醇与乙苯为底物,以分子氧为氧化剂,考察研究了固体催化剂的催化氧化性能。
1 实验部分
1.1试剂与仪器
CPS微球:交联度为4%,粒径为0.315~0.450 mm,常州市腾龙化工有限公司;1,4-二氯甲氧基丁烷(BCMB):自制;GA:化学纯,上海基丽化学技术有限公司;三乙胺(TEA):分析纯,西陇化工股份有限公司;间氨基苯酚(MAP):分析纯,上海科丰化学试剂有限公司;环己醇:分析纯,国药集团化学试剂有限公司;N,N-二甲基乙酰胺(DMAC):分析纯,国药集团化学试剂有限公司;N,N-二甲基甲酰胺(DMF):分析纯,青岛和兴精细化学有限公司;1,4-二氧六环(Dioxane):分析纯,青岛和兴精细化学有限公司;其余试剂均为市售分析纯。
美国Perk in-Elmer公司1700型傅里叶红外光谱仪,KBr压片法;S-4800型场发射扫描电子显微镜(SEM,日本日立株式会社);上海尤尼柯公司的UV-2602型紫外-可见分光光度计;GC-920型气相色谱仪(上海海欣色谱有限公司);UltiMate 3000型液相色谱仪(HPLC,C18反相色谱柱,美国Dionex公司)。
1.2在CPS微球表面固载氨基酚型双齿席夫碱氧钒(Ⅳ)配合物
1.2.1用乙醛酸改性CPS微球以自制的BCMB作为氯甲基化试剂,制得CMCPS微球(俗称为氯球),其中氯的质量分数为14%[23]。在装有电动搅拌器和冷凝回流管的100 mL四口瓶中,加入1 g氯球和50 mL溶剂DMAC,将微球溶胀12 h,使大分子链充分舒展。加入0.4 g GA,使之溶解,并加入1 mL TEA作为缚酸剂,置于90 ℃恒温水浴中,使氯球的氯甲基与GA的羧基之间发生酯化反应,8 h后结束反应。反应液静置,取一定量的上清液,用紫外分光光度法(λ=265 nm)测定上清液中剩余GA的浓度,从而计算参与反应的GA的量,进一步计算氯球表面氯甲基的转化率。抽滤并收集反应后产物微球,用蒸馏水和无水乙醇洗涤,40 ℃下恒温干燥24 h,即得表面键合有AG的改性微球CPS-AG。
1.2.2在CPS-AG微球表面键合氨基酚型双齿席夫碱配基在四口瓶中加入1.0 g改性微球CPS-AG和30 mL溶剂DMAC,使微球溶胀12 h,然后加入0.42 g的MAP,恒温90 ℃,在搅拌条件下使改性微球CPS-AG表面的AG和MAP分子中的伯氨基反应12 h,反应液静置,取一定量的上清液,用紫外分光光度法测定上清液中剩余MAP的浓度,从而间接测定CPS-AG表面醛基的转化率,测得转化率为84%。过滤,用蒸馏水和乙醇反复洗涤微球,40 ℃下恒温干燥24 h,即得功能微球CPS-AGAP。
1.2.3固载化席夫碱氧钒(Ⅳ)配合物催化剂的制备将2 g的硫酸氧钒充分溶解到40 mL溶剂DMAC中,再加入1 g CPS-AGAP微球,于80 ℃恒温,反应14 h,静置,取上清液样品,采用高锰酸钾氧化-硫酸亚铁铵滴定法(YB/T 5328-2009)测定配位反应后反应液中剩余的硫酸氧钒的量,间接测定出氧钒物中V=O在微球CPS-[VO(AGAP)2]表面的固载量为0.78 mmol/g[22]。收集产物微球,用蒸馏水与乙醇反复洗涤产物微球,抽滤,40 ℃下恒温干燥24 h,即得墨绿色的催化剂微球CPS-[VO(AGAP)2],其制备过程如图1所示。
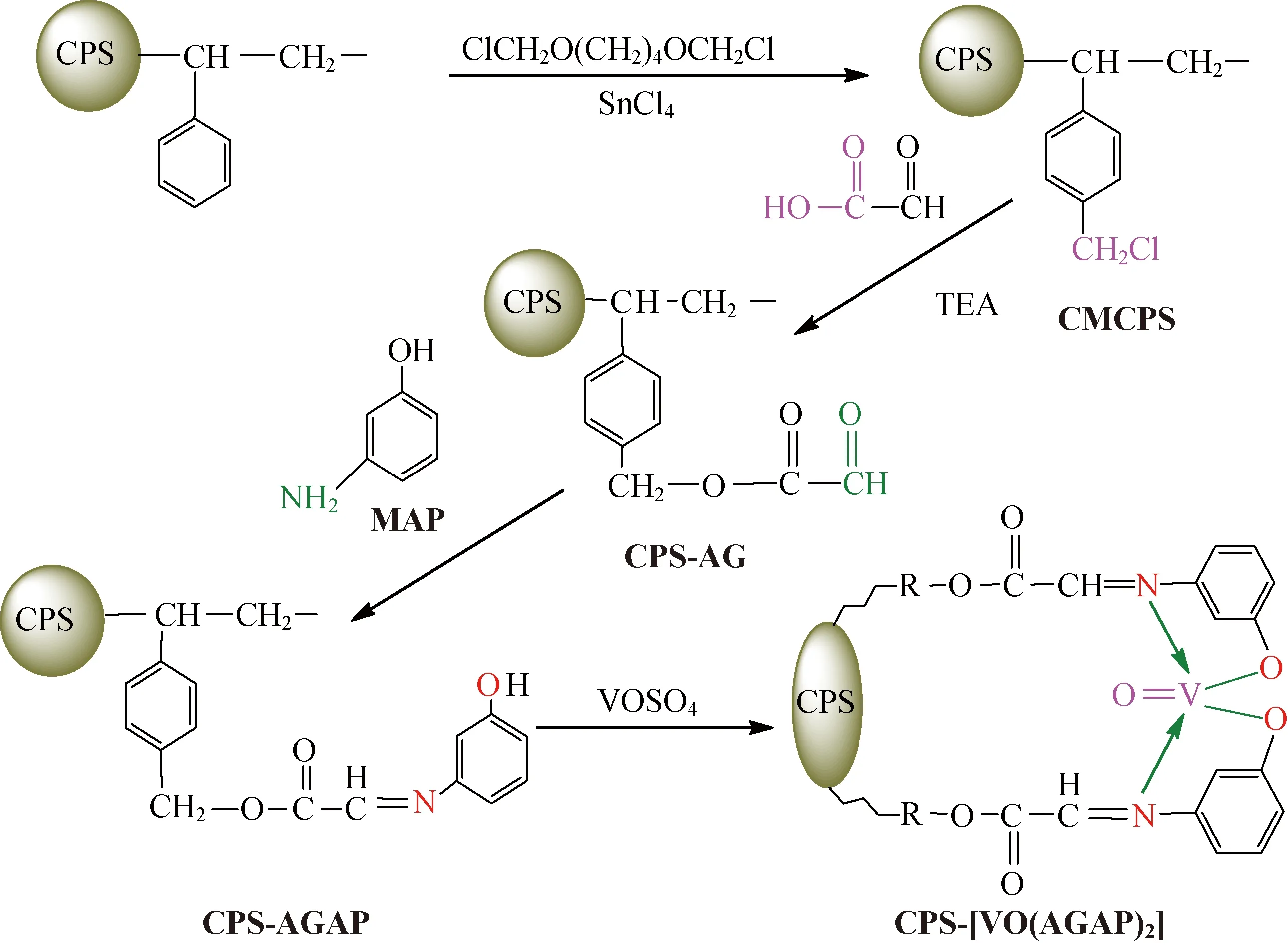
图1 制备催化剂微球CPS-[VO(AGAP)2]的化学反应过程
1.3以微球CPS-[VO(AGAP)2]为固体催化剂的分子氧催化氧化实验
1.3.1环己醇的分子氧催化氧化实验在装有电动搅拌器和冷凝管的100 mL四口瓶中,加入30 mL溶剂冰乙酸,再加入1 g催化剂微球CPS-[VO(AGAP)2],浸泡12 h,使微球充分溶胀,然后加入2 mL环己醇,常压下通入氧气且氧气的流速固定(用气体流量计控制),于80 ℃下恒温反应30 h。间隔一定时间取样,用GC-920型气相色谱仪(N2为载气,HP-5毛细管色谱柱,FID检测)分析产物,测定环己醇转化率。结果表明,在色谱谱线上只存在产物环己酮与底物环己醇的谱峰(保留时间分别为4.2 min 与5.0 min)。但在产物的紫外吸收光谱中,于210 nm处显现出极微小的己二酸吸收峰,故可初步推断,在本文体系中,环己醇分子氧催化氧化的主产物为环己酮,也含有极微量的己二酸。
1.3.2乙苯的分子氧催化氧化实验将1 g催化剂微球CPS-[VO(AGAP)2]加入到30 mL乙苯中,常压下通入氧气且氧气的流速固定(用气体流量计控制),90 ℃下恒温反应,每间隔一段时间取样,用高效液相色谱仪鉴定产物,测定乙苯的转化率。测定结果表明,苯乙酮几乎是唯一产物(色谱图中只出现了苯乙酮与乙苯的峰,保留时间分别为3.6 min 与5.8 min)。
2 结果与讨论
2.1催化剂微球CPS-[VO(AGAP)2]的表征
2.1.1红外光谱图2为氯球、改性微球CPS-AG、功能微球CPS-AGAP及催化剂微球CPS-[VO(AGAP)2]的红外光谱图。
在微球CMCPS的红外谱图中,1 445 cm-1和675 cm-1处的2个峰分别是氯甲基中C-H键的面内弯曲振动吸收峰和C-Cl键的伸缩振动吸收峰。但在改性微球CPS-AG的红外谱图中,这2个峰几乎消失,与此同时,在1 749 cm-1处出现了酯羰基的伸缩振动吸收,在1 628 cm-1处出现了醛基的特征吸收,在1 260 cm-1处出现酯基中C-O-C键的伸缩振动吸收峰。上述谱峰数据的变化充分表明,CMCPS已与GA发生了酯化反应,形成了键合有醛基的改性微球CPS-AG。
在功能微球CPS-AGAP的红外谱图中,1 628 cm-1处醛基的特征吸收已明显减弱或消失,同时在1 662 cm-1处出现了席夫碱反应后生成的亚胺C=N键的伸缩振动吸收,在3 385 cm-1处出现酚羟基的伸缩振动,表明在微球CPS表面已键合间氨基苯酚型双齿席夫碱配基,形成功能化微球CPS-AGAP。
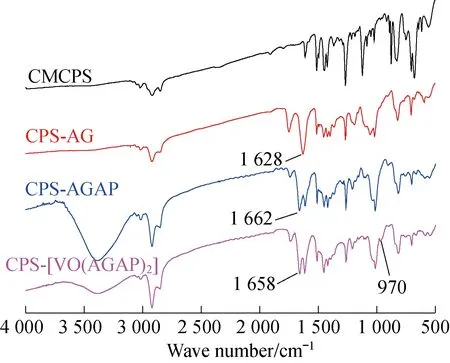
图2 4种微球的红外光谱
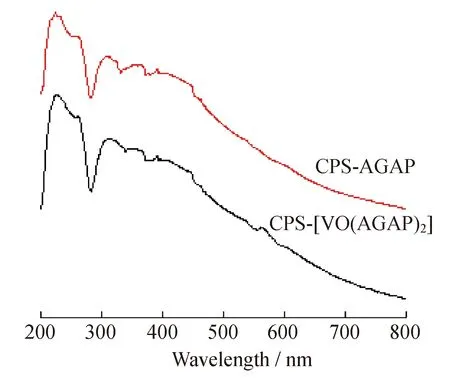
图3 微球CPS-AGAP与CPS-[VO(AGAP)2]的紫外-可见吸收光谱
在催化剂微球CPS-[VO(AGAP)2]的红外谱图中,于970 cm-1处出现V=O键的特征吸收峰;同时亚胺C=N键吸收峰红移4 cm-1(一般红移3~8 cm-1),标志着亚胺键N原子已与V原子发生配位;原酚羟基的特征吸收大为减弱,显示出酚氧原子也与V原子发生了配位,形成了键合在CPS微球表面的配合螯合物[VO(AGAP)2],即形成了固载氨基苯酚型双齿席夫碱氧钒配合物的催化剂微球CPS-[VO(AGAP)2]。总之,红外光谱数据基本表明在CPS微球表面已合成与固载了配合物[VO(AGAP)2],其化学结构将进一步被紫外-可见吸收光谱测试数据所证实。
2.1.2紫外-可见吸收光谱图3为功能微球CPS-AGAP与催化剂微球CPS-[VO(AGAP)2]的固体紫外吸收光谱。在微球CPS-AGAP的谱图中,存在3个吸收峰,于260 nm和309 nm处的2个吸收峰分别归属于苯环与亚胺键(C=N)的π→π*跃迁吸收;350 nm处的吸收峰归属于亚胺基团中N原子的n→π*跃迁吸收。在催化剂微球CPS-[VO(AGAP)2]的谱图中,上述3个吸收峰未发生实质性变化,表明键合配基AGAP的存在。但是,在可见光区域的564 nm处出现了由V原子d-d跃迁所引起的的吸收峰,表明VO2+离子已与键合配基发生配合反应。微球CPS-[VO(AGAP)2]的上述紫外-可见吸收光谱与其他氧钒配合物的十分相似[9,24]。故微球CPS-[VO(AGAP)2]的化学结构进一步被紫外-可见吸收光谱数据所证实。
2.1.3微球的形貌图4给出微球CPS与催化剂微球CPS-[VO(AGAP)2]的SEM照片。可以清楚地看出,微球CPS具有良好的球形度,且粒径均匀,表面光滑;微球CPS-[VO(AGAP)2]虽然仍保持着良好的球形度,但表面变得较为粗糙。

图4 微球CPS(a)与CPS-[VO(AGAP)2](b)的SEM照片
2.2主要因素对GA改性CPS微球反应的影响
在制备催化剂微球的4步反应中,CPS微球的氯甲基化反应技术成熟,醛基与伯氨基之间的席夫碱反应易于进行,双齿席夫碱配基与硫酸氧钒之间的配位反应在文献[21]中也曾实施过,因此,关键步骤是氯球CMCPS与GA之间的酯化反应,本文重点考察了主要因素对该反应的影响,从而优化了反应条件。
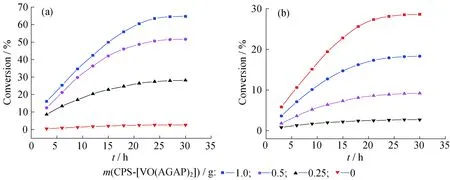
2.2.1溶剂极性的影响使用4种极性不同的溶剂:DMAC、DMF、DMF-Dioxane(体积比1∶1)混合溶剂和Dioxane,其介电常数依次为37.8、36.71、19.46、2.21,使微球CMCPS与GA在不同溶剂中发生酯化反应。图5为不同体系中微球 CMCPS氯甲基转化率随反应时间的变化曲线。从图5中可以看出,在其他反应条件相同的条件下,微球CMCPS表面的氯甲基与GA分子的羧基反应程度的顺序为:Dioxane 2.2.2反应温度的影响固定其他反应条件,不同温度下CMCPS的氯甲基转化率随反应时间的变化曲线如图6所示。在温度较低的时候,CMCPS微球表面的氯甲基与GA分子的羧基发生酯化反应效果较差,氯甲基的转化率较低;随着反应温度的不断升高,在相同的反应时间内氯甲基的转化率明显提高,符合反应动力学规律,温度升高,反应速率加快。90 ℃下恒温反应8 h,氯甲基转化率可以达到82%。实验中观察到,温度超过90 ℃,虽然CMCPS氯甲基的转化率会继续提高,但微球有破裂现象。可能的原因是进一步升高温度,使CMCPS微球的热稳定性变差。CPS微球的玻璃化转变温度在100 ℃附近,当温度接近或高于CPS微球的玻璃化转变温度时,构成微球的聚苯乙烯大分子链会发生断裂,导致微球破裂。因此,本文选择90 ℃为适宜的反应温度。 图5 使用不同溶剂时CMCPS微球中的氯甲基转化率随反应时间的变化曲线 图6 不同温度下CMCPS 的氯甲基转化率随反应时间的变化曲线 2.3催化剂微球CPS-[VO(AGAP)2]的催化氧化性能 2.3.1在分子氧氧化环己醇过程中的催化活性使用氧钒固载量为0.78 mmol/g的催化剂微球CPS-[VO(AGAP)2],以分子氧为氧化剂,使环己醇发生氧化反应,图7给出不同体系中环己醇转化率随反应时间的变化曲线。 图7 环己醇(a)和乙苯(b)转化率随反应时间的变化曲线 从图7(a)可以看出,不加催化剂的空白体系基本无反应发生;而加入催化剂微球CPS-[VO(AGAP)2]的反应体系,随反应时间延长,环己醇转化率大幅提高,充分显示了该微球对环己醇分子氧氧化反应的催化活性;同时,随着固体催化剂加入量的增大,相同反应时间内,环己醇的转化率在增大,表明反应速率在加快,体现出非均相催化反应的一般规律:在固体催化剂加入量的一定范围内,随着固体催化剂加入量的增大,化学反应速率加快。图7(a)显示,对于1.3.1节所述的反应体系,当加入1 g催化剂微球CPS-[VO(AGAP)2],反应30 h时,环己醇转化率可达64%。此结果表明,固载于聚合物微球表面的氨基酚型双齿席夫碱氧钒(Ⅳ)配合物,对于分子氧氧化环己醇的氧化反应具有良好的催化活性,可以在较为温和的条件(常压下的分子氧及不太高的温度)下实现环己醇向环己酮的氧化转化。氧钒(Ⅳ)配合物的催化氧化性能与钒原子具有互换的高价氧化态、拥有高配位数、处于氧化态的钒原子对氧分子具有高亲和力等密切相关,催化氧化机理中涉及到反应底物在钒原子周围的配位、过氧钒配合物的形成、过氧钒配合物从底物分子中提取氢等步骤[10,25],关于催化剂微球CPS-[VO(AGAP)2]在醇分子氧催化氧化过程中的催化机理还将进一步研究。 2.3.2在分子氧氧化乙苯过程中的催化活性仍使用氧钒固载量为0.78 mmol/g的催化剂微球CPS-[VO(AGAP)2],以分子氧为氧化剂,使乙苯发生氧化反应,图7(b)给出不同体系中乙苯转化率随反应时间的变化曲线。 图7(b)显示,不加催化剂的空白体系基本无反应发生;而加入催化剂微球CPS-[VO(AGAP)2]的反应体系,随反应时间延长,乙苯转化率在不断提高,显示出该催化剂微球对乙苯的分子氧氧化反应也有明显的催化活性;随着固体催化剂加入量的增大,相同反应时间内,乙苯的转化率在增大,说明反应速率在加快。图7(b)显示,对于1.3.2节所述的反应体系,当加入1 g催化剂微球CPS-[VO(AGAP)2],反应30 h时,乙苯转化率为29%。此实验结果表明,固载于聚合物微球表面的氨基酚型双齿席夫碱氧钒(Ⅳ)配合物,不仅可催化分子氧氧化醇类物质的氧化反应,将醇类物质转变为相应的羰基化合物,而且也能催化烷基芳烃的氧化反应,其根本原因仍在于如上所述的氧钒(Ⅳ)物种的若干特点。在氧钒(Ⅳ)配合物催化乙苯(也包括碳氢化合物)的分子氧催化氧化过程中,氧化反应遵循自由基反应历程,其重要的中间产物为氢过氧化物,在氧钒(Ⅳ)配合物的催化作用下,该中间产物进一步转化为苯乙酮[26-27]。关于微球CPS-[VO(AGAP)2]对乙苯的催化性能和催化氧化机理有待进行更深入地研究。 (1) 以CPS微球为载体,通过多步大分子反应,在CPS微球表面同步合成与键合了氨基酚型双齿席夫碱配基,又经过与硫酸氧钒(Ⅳ)的配位螯合反应,制得了固载化的氨基酚型双齿席夫碱氧钒(Ⅳ)配合物微球CPS-[VO(AGAP)2]。其中,GA与GMCPS微球之间的酯化反应是关键步骤。 (2) 该酯化反应属亲核取代反应,使用极性强的溶剂有利于氯甲基C-Cl键的断裂,即有利于苄基碳正离子的形成,可加速反应的进行。 (3) 酯化反应适宜的反应溶剂为N,N-二甲基乙酰胺,适宜的反应温度为90 ℃,此时,CMCPS微球氯甲基的转化率可达82%。 (4) 在分子氧氧化环己醇为环己酮和分子氧氧化乙苯为苯乙酮的氧化反应中,微球CPS-[VO(AGAP)2]均具有良好的催化活性,均可在温和条件下(常压下的氧气和低于100 ℃的温度下)有效地实现两种有机物的氧化转变。 [1]HABER J,MATACHAWSKI L,PAMIN K,etal.Supported polyhalogenated metalloporphyrins as catalysts for the oxidation of cycloalkanes with molecular oxygen in lyons system[J].Catalysis Today,2004,91-92(s4):195-198. [2]SALAVATI-NIASARI M.Synthesis,characterization and catalytic oxidation of cyclohexene with molecular oxygen with host (nanopores of zeolite-Y)/guest (Ni(Ⅱ) complexes of 14-and 16-membered tetraaza dioxo diphenyl macrocyclic ligands) nanocomposite materials[J].Polyhedron,2008,27(14):3132-3140. [3]AHMAD J U,FIGIEL P J,RAISANEN M T,etal.Aerobic oxidation of benzylic alcohols with bis(3,5-di-tert-butylsalicylaldimine)copper(Ⅱ) complexes[J].Applied Catalysis A:General,2009,371(1):17-21. [4]NAIK R,JOSHI P,DESHPANDE R K.Immobilization of metallporphyrins on polystyrene:Efficient catalysts for aerobic oxidation of alcohols[J].Journal of Molecular Catalysis A:Chemical,2005,238(1-2):46-50. [5]GOIFMAN A,GUN J,GITIS V,etal.Pyrolysed carbon supported cobalt porphyrin:A potent catalyst for oxidation of hydrogen sulfide[J].Applied Catalysis B:Environmental,2004,54(4):225-235. [6]DHAKSHINAMOORTHY A,PITCHUMANI K.Clay-supported ceric ammonium nitrate as an effective,viable catalyst in the oxidation of olefins,chalcones and sulfides by molecular oxygen[J].Catalysis Communications,2009,10(6):872-878. [7]HABIBI D,FARAJI A R,ARSHADI M,etal.Manganese nanocatalyst andN-hydroxyphthalimide as an efficient catalytic system for selective oxidation of ethylbenzene,cyclohexene and oximes under aerobic condition[J].Journal of Molecular Catalysis A:Chemical,2014,382(4):41-54. [8]ISLAM S M,MOLLA R A,ROY A S,etal.Chem inform abstract:Aerobic oxidation and oxidative bromination in aqueous medium using polymer anchored oxovanadium complex[J].Journal of Organometallic Chemistry,2014,761(45):169-178. [9]MARCHETTI F,PETTINARI C,NICOLA C D,etal.Synthesis and characterization of novel oxovanadium(Ⅳ) complexes with 4-acyl-5-pyrazolone donor ligands:Evaluation of their catalytic activity for the oxidation of styrene derivatives[J].Applied Catalysis A:General,2010,378(2):211-220. [10]SILVA J A L D,SILVA J J R F D,POMBEIRO A J L.Oxovanadium complexes in catalytic oxidations[J].Coordination Chemistry Reviews,2011,255(19):2232-2248. [11]GRIVANI G,BRUNO G,RUDBARI H A,etal.Synthesis,characterization and crystal structure determination of a new oxovanadium(Ⅳ) Schiff base complex:The catalytic activity in the epoxidation of cyclooctene[J].Inorganic Chemistry Communications,2012,18(11):15-20. [12]MOHEBBI S,NIKPOUR F,RAIATI S.Homogeneous green catalyst for epoxidation of cyclooctene by mono oxovanadium(Ⅳ) complexes of N2O2donate ligand system[J].Journal of Molecular Catalysis A:Chemical,2006,256(1-2):265-268. [13]LOBMAIER G M,TRAUTHWEIN H,FREY G D,etal.Oxovanadium(Ⅳ) complexes as molecular catalysts in epoxidation:Simple access to pyridylalkoxide derivatives[J].Journal of Organometallic Chemistry,2006,691(10):2291-2296. [14]MAURYA M R,ARYA A,ADAO P,etal.Immobilisation of oxovanadium(Ⅳ),dioxomolybdenum(Ⅵ) and copper(Ⅱ) complexes on polymers for the oxidation of styrene,cyclohexene and ethylbenzene[J].Applied Catalysis A:General,2008,351 (2):239-252. [15]PEREIRA C,BIERNACKI K,REBELO S L H,etal.Designing heterogeneous oxovanadium and copper acetylacetonate catalysts:Effect of covalent immobilisation in epoxidation and aziridination reactions[J].Journal of Molecular Catalysis A:Chemical,2009,312(312):53-64. [16]MAURYA M R,CHANDRAKAR A K,CH S.Oxidation of methyl phenyl sulfide,diphenyl sulfide and styrene by oxovanadium(Ⅳ) and copper(Ⅱ) complexes of NS donor ligand encapsulated in zeolite-Y[J].Journal of Molecular Catalysis A:Chemical,2007,278(1):12-21. [17]PEREIRA C,BIERNACKI K,REBELO S L H,etal.Designing heterogeneous oxovanadium and copper acetylacetonate catalysts:Effect of covalent immobilisation in epoxidation and aziridination reactions[J].Journal of Molecular Catalysis A:Chemical,2009,312(312):53-64. [18]MAURYA M R,SIKARWAR S,KUMAR M.Oxovanadium(Ⅳ) complex ofβ-alanine derived ligand immobilised on polystyrene for the oxidation of various organic substrates[J].Catalysis Communications,2007,8(12):2017-2024. [19]BO F,YU H C,HUANG J W,etal.Mn(Ⅲ) porphyrins immobilized on magnetic polymer nanospheres as biomimetic catalysts hydroxylating cyclohexane with molecular oxygen[J].Journal of Molecular Catalysis A:Chemical,2009,298(1-2):74-80. [20]GUPTA K C,SUTAR A K,LIN C C.Polymer-supported Schiff base complexes in oxidation reactions[J].Coordination Chemistry Reviews,2009,253(13-14):1926-1946. [21]GAO B J,LI Y B,SHI N.Oxovanadium (Ⅳ) Schiff base complex immobilized on CPS microspheres as heterogeneous catalyst for aerobic selective oxidation of ethyl benzene to acetophenone[J].Reactive and Functional Polymers,2013,73(11):1573-1579. [22]李艳飞,高保娇,余依玲.固载化N,N-双齿席夫碱型氧钒(Ⅳ)配合物的制备及其在分子氧氧化乙苯中的催化作用[J].分子催化,2013,27(3):271-278. [23]申艳玲,杨云峰,高保娇.制备氯甲基化聚苯乙烯交联微球的新方法[J].高等学校化学学报,2007,28(3):580-583. [24]RAYATI S,KOLIAEI M,ASHOURI F,etal.Oxovanadium(Ⅳ) Schiff base complexes derived from 2,2′-dimethylpropandiamine:A homogeneous catalyst for cyclooctene and styrene oxidation[J].Applied Catalysis A:General,2008,346(s 1-2):65-71. [25]REDDY S R,DAS S,PUNNIYAMURTHY T.Polyaniline-supported vanadium catalyzed aerobic oxidation of alcohols to aldehydes and ketones[J].Cheminform,2004,35(34):3561-3564. [26]MISHRA G S,POMBEIRO A J L.Oxyfunctionalization ofn-pentane andn-hexane by oxovanadium complexes supported on carbamated modified silica gel[J].Applied Catalysis A:General,2006,304(1):185-194. [27]MISHRA G S,POMBEIRO A J L.Selective single-pot oxidation of cyclohexane by molecular oxygen in presence of bis(maltolato)oxovanadium complexes covalently bonded to carbamated modified silica gel[J].Journal of Molecular Catalysis A:Chemical,2005,239(1-2):96-102. Immobilizing Aminophenol Type Bidentate Schiff Base Oxovanadium(Ⅳ) Complex on Surfaces of CPS Microspheres and Its Catalytic Oxidation Property MENG Su-qing,CUI Kun-li,GAO Bao-jiao (Department of Chemical Engineering,North University of China,Taiyuan 030051,China) Esterification reaction between glyoxylic acid (GA) and chloromethylated crosslinked polystyrene (CMCPS) microspheres was made to be carried out,and the aldehyde group (AG) was introduced onto the surfaces of crosslinked polystyrene (CPS) microspheres,resulting in the modified microspheres CPS-AG.Subsequently,the Schiff base reaction of the AG on CPS-AG microspheres was conducted usingm-aminophenol as reagent,obtaining the functional microspheres CPS-AGAP,on which aminophenol type bidentate Schiff base ligands were bonded.Finally,the chelating coordination reaction between CPS-AGAP microspheres and vanadyl sulfate was performed,and microspheres CPS-[VO(AGAP)2],on which aminophenol type bidentate Schiff base oxovanadium(Ⅳ) complex was immobilized,were successfully prepared.The effects of the main factors on the esterification reaction between GA and CMCPS microspheres were studied and the reaction conditions were optimized.The microspheres CPS-[VO(AGAP)2] were fully characterized by Fourier Transform Infrared spectroscopy (FT-IR),Ultra-Violet spectrum (UV) and Scanning Electron Microscope (SEM).The microspheres CPS-[VO(AGAP)2] were used in the oxidations of cyclohexanol and ethylbenzene with molecular oxygen as oxidant,respectively,and their catalytic activity was examined.Results showed that the polarity of the solvents was advantageous to the esterification reaction between GA and CMCPS microspheres,and the suitable solvent wasN,N-dimethylacetamide with strong polarity.The appropriate reaction temperature was 90 ℃.Under the optimum reaction conditions,the conversion of the chloromethyl group of CMCPS microspheres could reach 82%.In the aerobic oxidations of cyclohexanol and ethylbenzene,CPS-[VO(AGAP)2] microspheres exhibited fine catalytic activity. crosslinked polystyrene microspheres; bidentate Schiff base ligand; oxovanadium(Ⅳ) complex; immobilization; catalytic oxidation 1008-9357(2016)03-0305-008 10.14133/j.cnki.1008-9357.2016.03.008 2016-04-22 山西省青年基金(2014021015-1) 孟素青(1990-),女,山东滨州人,硕士生,主要研究方向为功能高分子材料。E-mail:18734913259@163.com 高保娇,E-mail:gaobaojiao@126.com O631 A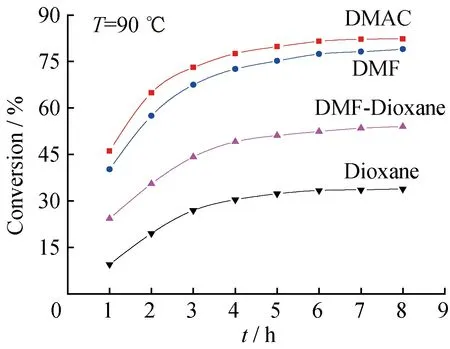
3 结 论
