超高效液相色谱-三重四级杆串联质谱法同时测定植物组织中多种激素
2016-10-16龚明霞王日升何龙飞
龚明霞,王日升,何龙飞,王 萌, 赵 虎,吴 星,何 志
(1.广西农科院蔬菜研究所,广西南宁 530007;2.广西大学农学院,广西南宁 530004)
植物激素主要包括细胞分裂素、生长素、赤霉素、脱落酸、茉莉酸等。它们广泛分布在植物体内,参与植物的生长、发育及各种生物胁迫和非生物胁迫过程[1,2]。如生长素-赤霉素类在器官生长[3]、生长素和脱落酸在热诱导的向下生长[4]、生长素、脱落酸、细胞分裂素、赤霉素、水杨酸及茉莉酸在盐胁迫响应[5]等过程中的调控。因此,对多种植物激素同时进行定量十分必要。
植物激素在植物体内的含量极低,通常只有普通植物次生代谢产物的万分之一甚至更低[6],因此,需要建立微量植物激素灵敏的测定方法。在对植物激素进行定量分析方面,早期有酶联免疫吸附法(ELISA)[7]、高效液相色谱法(HPLC)[8]、气相色谱法(GC)[9]、毛细管电泳法(CE)[10]等方法。但这些方法对植物内源激素的准确定性和定量存在某些方面不足。高效液相色谱-串联质谱法(HPLC-MS/MS)灵敏度高,选择性和特异性好,抗干扰能力强,不需要进行衍生化,可同时检测植物体内 3种以上内源激素[11,12]。然而,对植物体内几大类天然存在的、主要作用形式的几种激素如反式玉米素(Trans-ZT)、吲哚乙酸(IAA)、天然脱落酸(S-ABA)、赤霉素类(GA1、GA3、GA4)和茉莉酸(JA)的同时定量研究还未见报道。本文研究的7种激素的结构见图1。
近年来,随着液相色谱柱填充物的不断发展,超高效液相色谱(UHPLC)越来越普及,与传统液相色谱柱相比,在分离效率、分辨率、灵敏度、分析速度和样品数量方面均有所改进[13,14],且有较好的选择性保留性能,可以将基质效应降到最小[15]。超高效液相色谱-三重四级杆串联质谱(UHPLC-QqQ-MS/MS)采用选择性更高的多反应监测(MRM)模式,能进一步减少或消除基质的干扰,即使在复杂基质样品中也能得到更低的检测限。本研究拟利用辣椒叶片为基质,提取和纯化5大类植物内源激素中的7种,即ZT、IAA、ABA、GA1、GA3、GA4、JA进行色谱和质谱条件优化,旨在提出一种能够对多种植物激素同时提取和准确定量的UHPLC-MS/MS方法。
1 实验部分
1.1 主要仪器与试剂
Agilent 1290 Infinity色谱系统,Agilent 6460 Triple Quad三重四级杆质谱仪(美国);Eppendorf5430R和5810R型台式冷冻离心机(德国);Grant XUB25型超声波仪(英国);EYELA旋转减压蒸发仪(日本);SUPELC 57265型固相萃取装置(美国);Martin Christ ALPHA 1-4 LD型冷冻干燥机(德国)。
7种标准品:Trans-玉米素(Trans-TZ)、赤毒素(GA1和GA4)、脱落酸(ABA)、茉莉酸[(±)JA]购于捷克Olchemim Ltd.,赤霉酸(GA3)和吲哚乙酸(IAA)购于德国Dr.Ehrenstorfer GmbH公司。各标准品用甲醇溶解,配制成1 mg·mL-1的单标储备液,并置于-40 ℃冰箱中密封保存。采用甲醇逐级稀释,配制系列混合标准工作液:IAA的浓度梯度为0.4 、5、50、200、500 ng·mL-1,ZT的浓度梯度为0.05、0.5、5、50、200 ng·mL-1,ABA的浓度梯度为15、50、100、200、500 ng·mL-1,GA1、GA3、GA4及JA的浓度梯度均为50、100、200、400、500 ng·mL-1。配制3个加标混合液,ZT、IAA的浓度梯度均为2、20、100 ng·mL-1,GA1、GA3、GA4、JA、ABA的浓度梯度均为60、150、300 ng·mL-1。甲醇、乙腈(Merck,德国),甲酸(TEDIA,美国)均为色谱纯。
1.2 样品处理
1.2.1提取植物组织加液氮研磨后,准确称取0.5 g(精确至0.001 g)于10 mL离心管中,加入5 mL经4 ℃预冷的改良型的Bieleski试剂(甲醇∶甲酸∶水=15∶1∶4),于4 ℃下浸提16 h,期间振荡多次,4 ℃ 12 000 r/min离心15 min得上清液,再向残渣中加入4 mL预冷的提取液,振荡提取2 h后离心获得上清液;重复提取2次,合并3次上清液;在38 ℃恒温水浴中旋转减压蒸发至水相,置-20 ℃冰箱中冷冻30 min。
1.2.2纯化从-20 ℃冰箱取出解冻,4 ℃ 12 000 r/min离心10 min,取上清液,弃色素及脂类不溶物;用1 mol/L HCl调pH值为3.0,过C18-SPE小柱(规格200 mg/3mL,Agilent),液体在自然重力下流出。C18-SPE柱使用前采用3 mL 100%甲醇活化和3 mL 0.1 mol/L甲酸水溶液平衡。用3 mL 0.1 mol/L甲酸水溶液淋洗1次,并减压抽干,接着用3 mL 70%甲醇洗脱,收集洗脱液,38 ℃旋转减压蒸发至水相后,置冷冻干燥机中进行冷冻干燥,用300 μL甲醇溶解干燥残渣(低温下超声波助溶30~60 s),然后过0.22 μm有机系滤膜,供UHPLC-QqQ-MS/MS测定。
1.3 色谱-质谱条件
色谱条件:Agilent ZORBAX RRHD Eclipse Plus C18色谱柱(50×2.1 mm,1.8 μm)。以甲醇(A)和含0.1% 甲酸水溶液(B)为流动相。梯度洗脱程序:0~2 min,35%A;2~6 min,35%~45%A;6~9 min,45%~50%A;9~15 min,50%~60%A;15~18 min,60%~100%A;18~20 min,100%~35%A;20~22 min,35%A。流速:0.3 mL/min;上样量:1 μL。
质谱条件:离子源为安捷伦6460 QQQ喷射流离子聚焦电喷雾源,采用正、负离子快速切换,多反应监测(MRM)模式监测;干燥气体为N2,温度为300 ℃,流速为10 L/min;雾化气体为N2,雾化气压力为2.8×105Pa;鞘气气体为N2,温度为360 ℃,流速为12 L/min;毛细管电压为4 000 V(+)或3 500 V(-),喷嘴电压为0 V。
1.4 定性与定量分析
对分析样品溶液进行UHPLC-QqQ-MS/MS测定,以各种激素的保留时间和特征离子对为依据进行定性,采用外标法以定量离子对的峰面积进行定量。
2 结果与讨论
2.1 色谱条件的优化
首先,分析目标物的结构和极性,ZT、IAA、GA1、GA3、GA4、ABA、JA这7种植物激素的分子结构中均包含疏水性的基团,所以我们选择反相C18色谱柱来对它们进行分离。ZT分子中嘌呤环侧链末端带一个极性羟基,使得整个分子的极性偏强;IAA分子中碳链末端带有一个极性羧基;GAs分子中弱极性的碳环被极性的羟其、羧基和酯基包围而使得整个分子具有中等极性;ABA和JA分子中均带有极性的羟基和羧基。可见,7种植物激素的极性存在差异,经优化选择了1.3所述的梯度洗脱条件。
7种分析物中大部分是酸性植物激素,为了使它们能够在反相色谱柱上充分保留,采用0.1% 甲酸水溶液为无机相,分别考察了乙腈、甲醇作为有机相对这7种植物激素的分离效果。结果表明:以乙腈-0.1% 甲酸水溶液作为流动相,7种激素几乎在0~3 min内出峰,但有些峰重叠,说明不能完全分离;以甲醇-0.1% 甲酸水溶液作为流动相,各激素的出峰时间延迟,但都能在14 min之内实现基线分离,没有重叠峰,无拖尾现象,而且峰形好。
2.2 质谱条件的优化
赤霉素、脱落酸、茉莉酸这三大类化合物分子中均含有羧基,在电离源中易产生[M-H]-离子[15],因此宜采用负离子模式。对1 μg/mL各激素单标溶液进行母离子全扫描,结果也表明GA1、GA3、GA4、ABA、JA在负离子模式下响应值高,而ZT和IAA在正离子模式下响应值高;且可以看到对相同质量浓度的单标溶液,正离子模式下的ZT和IAA的响应值远高于负离子模式下的GA1、GA3、GA4、ABA、JA,充分说明了分子结构不同的7种植物激素的离子化效率也不一样[16],Fletcher和Mader的研究结果表明碱性添加物有利于增强分析物在负离子模式下的离子化效率[17]。因此,本研究结果可进一步推测负离子模式下使用酸性流动相可能会抑制分析物的离子化,正离子模式下使用酸性流动相可能会促进分析物的离子化。
通过对各种激素母离子进行二次碎裂,得到二级质谱特征离子谱图,从中选出各物质最强且稳定的子离子作为定量子离子。ZT、IAA的准分子离子峰为[M+H]+,GA1、GA3、GA4、ABA、JA的准分子离子峰为[M-H]-。进行正、负离子模式切换多反应监测时,分别对ZTm/z136.0、IAAm/z130.0、GA1m/z273.1、GA3m/z143.2、GA4m/z257.1、ABAm/z153.1、JAm/z58.9的碎片离子进行监测。再优化其他质谱条件,使准分子离子→子离子的响应强度达到最大。表1为优化的源内碎裂电压和碰撞能量。

表1 优化的质谱分析参数
2.3 C18-SPE柱的萃取效果
尽管植物粗提液中的激素也能被定量检测,但对其进行纯化处理后再检测可大大提高LC-MS/MS系统中色谱柱的寿命[18],同时也极大地减少了植物基质中杂质对分析物离子化的影响[19]。C18柱是使用最为广泛的一种样品处理小柱。采用5份相同的含中等浓度7种植物激素的混合标样300 μL,加入与处理0.5 g植物组织相同量的提取液,直接进行旋转减压蒸发至水相,用1 mol/L HCl调pH值为3.0,过C18-SPE小柱(规格200 mg/3mL,Agilent),之后的步骤如同前面提到的。如表2所示:7种植物激素的混合标样经C18-SPE柱萃取后的回收率为92.2%~99.8%,相对标准偏差(RSD,n=5)为1.1%~2.1%,这说明C18-SPE柱能够有效吸附这7种激素,5 mL 70%甲醇能够完全洗脱这7种激素。另外,辣椒叶片样品粗提液,经C18-SPE小柱萃取时,可以见到大部分的有色杂质不能保留在柱内,直接随废液流出,而最终制备得到的检测液接近无色,这说明C18-SPE柱对糖类、酚类、色素等强极性杂质不吸附或弱吸附,使得它们在通过C18-SPE柱时直接流出或在淋洗过程被洗脱,从而能够有效去除样本基质中大部分的干扰杂质。

表2 7种植物激素经C18-SPE柱萃取后的回收率和相对标准偏差(n=5)
2.4 线性关系、检出限以及定量限
将系列混合标样溶液过0.22 μm滤膜后进行UHPLC-QqQ-MS/MS分析,每个样进5针。以峰面积(y)为纵坐标、质量浓度(x)为横坐标制作标准曲线,进行线性回归,并计算方法的检出限(LOD)(信噪比S/N=3) 和定量限(LOQ)(信噪比S/N=10)。从表3可以看出,7种植物激素在所采用的浓度范围内与其峰面积呈良好的线性关系,相关系数(R2)范围为0.9870~0.9990,且该方法检测的灵敏度高,检出限在0.01~8.77 ng·mL-1之间,定量限在0.02~29.23 ng·mL-1之间。
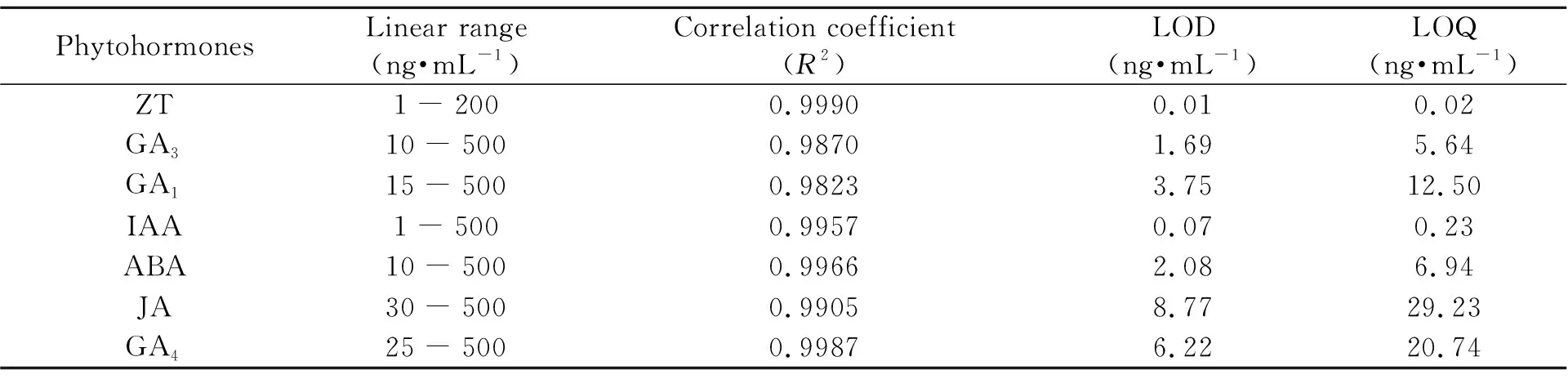
表3 7种植物激素的线性范围、相关系数、检出限和定量限
2.5 方法的回收率和精密度
以新鲜辣椒叶片为植物材料。准确称取同一辣椒叶片样品20份,每份0.5 g,其中15份加入低、中、高3个浓度水平的混合标样300 μL,每个水平设5次平行实验,另外5份为未加标的样品。所有样品按照前述的样品前处理方法进行提取、纯化和UHPLC-MS/MS检测。结果见表4。采用改良型的Bieleski试剂作为提取液,在3种不同添加水平下,辣椒叶片基质中的7种植物激素的回收率在65.8%~90.9%之间,大部分的回收率都较高,如GAs、ABA及JA的回收率均在82%以上,只有ZT的回收率相对偏低,相对标准偏差(RSD)为2.1%~5.5%。加标的辣椒叶片样品的MRM提取色谱图见图2。

表4 7种植物激素在辣椒叶片组织中的加标回收率和相对标准偏差(n=5)
植物激素回收率的高低与提取液成分有很大的关系,改良型的Bieleski试剂已被广泛应用于细胞分裂素、脱落酸、赤霉素、生长素等多种植物激素的提取[20]。本研究采用改良型的Bieleski试剂作提取液,冷冻离心结合C18-SPE柱萃取去杂纯化步骤,对大部分植物激素种类而言能获得较高的回收率,只有ZT的回收率偏低,与前人的研究结果大体一致,但在回收率上又有很大的提高。
3 结论
本研究建立了超高效液相色谱-三重四级杆串联质谱同时测定五大类植物激素中的7种即ZT、IAA、ABA、GA1、GA3、GA4、JA的分析方法。在优化的实验条件下,7种激素用Agilent ZORBAX RRHD Eclipse Plus C18色谱柱分离后,在QqQ-MS/MSMRM模式下进行定性和定量分析。该方法分析一个样品只需要14 min,检测激素种类范围广泛,且灵敏度高。植物样本前处理采用改良型的Bieleski试剂提取,结合C18-SPE小柱对植物样品进行富集和纯化,降低了基质干扰,研究中所分析的大部分植物激素的回收率都较高,适用于对植物组织中多种激素的同时测定。
