Overall Evolutionary Relationship of Cellulases from Archaea,Bacteria and Eukaryota*
2016-10-13严少敏
严少敏,吴 光
(广西科学院,非粮生物质酶解国家重点实验室,国家非粮生物质能源工程技术研究中心,广西生物质产业化工程院,广西生物炼制重点实验室,广西生物科学与技术研究中心,广西南宁 530007)
Overall Evolutionary Relationship of Cellulases from Archaea,Bacteria and Eukaryota*
严少敏,吴光
(广西科学院,非粮生物质酶解国家重点实验室,国家非粮生物质能源工程技术研究中心,广西生物质产业化工程院,广西生物炼制重点实验室,广西生物科学与技术研究中心,广西南宁530007)
【Objective】Cellulases have wide applications in biofuel,food,paper,pharmaceutical,and textile industries.Many organisms produce cellulases,therefore there is a very complicated evolutionary relationship for cellulases.Such a relationship is further tangled by the efforts that manipulate cellulase structures and express cellulase genes in different species to improve their activity and suitability.So far,no large-scale phylogenetic studies have been conducted to analyze the overall evolutionary relationship of cellulases.Yet no statistical studies have been conducted to exhaustively analyze variances of cellulases and their partitioned variances along taxonomic lineage from superkingdom to organism.【Methods】To fill these knowledge gaps,2 416 cellulases,including 116,1 744 and 556 from archaea,bacteria and eukaryota,were studied using statistical and phylogenetic analyses.Statistical analysis was conducted by computing the average pairwise p-distance for each phylum,class,order,family and genus,and by further partitioning the variance into inter-clan variance and intra-clan variance along taxonomic lineage.Large-scale phylogenetic analysis was performed through the phylogenetic tree of cellulases.【Results】The average pairwise p- distance of amino acid sequences varied greatly in the cellulases from different phylum,class,order,family and genus.The inter-clan variance and intra-clan variance varied also greatly along taxonomic lineage but a light pattern could be found in eukaryota.For large-scale phylogenetic analyses,most cellulases underwent different paths and bifurcations,and formed intertwined clusters,where many meaningful hints can be tracked.【Conclusion】This large-scale study provides the insights on overall evolutionary relationship of cellulases from both statistical and phylogenetic viewpoints.As additional information,the statistical analysis not only enriches the phylogenetic analysis,but also suggests how to biotechnologically manipulate cellulases with desired qualities,and how the evolution of cellulases are involved with vertical inheritance,horizontal gene transfer,multiple times and multiple clans.
cellulase,evolution,inter-clan variance,intra-clan variance,phylogenetics
0 Background
【Significance】Cellulases are a suite of enzymes catalyzing endohydrolysis of 1,4-beta-D-glucosidic linkages in cellulose,lichenin and cereal beta-D-glucans,which are the components of biomass,and have wide applications in biotechnology industries ranging across food,textile,paper,and pharmaceutical industries[1].Over last years,great attention is given to cellulases’ potential application in biofuel industry because they play a crucial role in the processing of enzymatic conversion from biomass to ethanol[2].【Previous progress】Huge efforts have been given to enhance cellulase’s activity[3-5]and to improve their pH optimum[6],thermostability[7-8],etc.Of many focuses in either academic or biotechnological settings,man-made mutations were introduced in order to modify the reaction center in cellulases[9-10],modify the carbohydrate-binding module (CBM) in cellulases[11],for example.Thereafter,all the positive structure modifications are desired to keep through generations of the organisms that produce cellulases[11].The manipulations of cellulases are done through experimental approaches with the hope that any manipulations in cellulases can pass the evolutionary test.Many cellulase variants can survive from generation to generation,however many of them are short-lived,and cellulases have different tendencies in maintaining their designed advantages in different species,i.e.the so-called strain-specific cellulase[12].Indeed,many cellulolytic microorganisms can produce cellulases[13-14],including bacteria,fungi,protozoans,and some animal species,such as termites and crayfish[15-16].Nevertheless,the primary resources are related to archaea,bacteria and eukaryota,which account for the most part of cellulases used in research and industry[17].【Insertion point】The wide existence of cellulases implicates a very complicated evolutionary relationship for all the organisms that produce cellulases because the Darwinian theory suggests that a common ancestor should have existed at the very beginning[18].As a result,the knowledge on the overall evolutionary relationship of cellulases would be necessarily useful[19-22].Early observations showed that a large taxonomic clan would have more variants than a small taxonomic clan does[18],which statistically suggests the importance of knowing the variance in each clan.This is because a mutant would have a large chance to survive if it comes from a clan whose variance is statistically large.For this reason,it is important to conduct statistical analysis in each clan of cellulases in order to find out their variance,which in fact is the average pairwise p-distance in each clan[23].This result would give the advantage to identify the clan,where a manipulated mutant easily survives.From an evolutionary viewpoint,the larger the average pairwise p-distance in a clan is,the stronger the selective pressure would be,and the larger the probability of evolution in multiple times is.Naturally,the computation of the average pairwise p-distance in each clan requires the cellulases that have clearly and fully documented taxonomic lineage from superkingdom to kingdom,to phylum,to class,to order,to family,to genus and to organism.【Key issues to be resolved】It is often necessary to express a cellulase gene in a species that can adapt to unfavorable environments because this gene may have a high productivity of cellulase but its original species is hard to adapt to unfavorable environments[24].This is the question of whether a mutant can survive across clans.Statistically,it is not enough to just know the variance within each clan,but it is also equally important to know the variance between clans,namely,to partition the variance of p-distance into between clans and within clan.This is because a mutant would have a large chance to survive in another species if the variance between two species is large,i.e.the variance is so large that two species could be mixed together[25].So it is necessary to compute inter- (between) and intra- (within) taxonomic clan variance using model Ⅱ ANOVA[26-32],and this variance is actually based on p-distance,which marks diversity.This result would also give the advantage to identify the clan,where a manipulated mutant easily survives and its surviving environments are more econsomically accessible.From an evolutionary viewpoint,the larger the inter-clan variance is,the larger the probability of horizontal gene transfer is,while the larger the intra-clan variance is,the larger the probability of vertical inherence is,the large the probability of survival of lifestyle-specific genes is[33].Similarly,this computation requires the cellulases that have clearly and fully documented taxonomic lineage including superkingdom,kingdom,phylum,class,order,family,genus and organism.
Furthermore,a large-scale phylogenetic analysis on cellulases is necessary to improve our understanding of their overall evolutionary relationship because the evolutionary roadmap can be tracked step-by-step from a phylogenetic tree[34-35].As cellulases are found in all three superkingdoms,archaea,eukaryota and bacteria,their overall phylogenetic tree can reveal the occurrence of their separation events and their further evolution and multiple evolutionary signals as well.Indeed,these events lead to cellulases to evolve independently
multiple times into multiple clans.Basically,multiple clan evolution in each superkingdom is more visible when phylogentic trees are made with respect to archaea,eukaryota and bacteria separately.In this context,it is possible to define who is responsible for the horizontal gene transfer because the horizontal gene transfer of cellulase was observed in 1998[36]and horizontal transfers of genetic materials of cellulase across clan were furthermore found[37-38].Phylogenetic reconstructions also would reveal whether a single gene was present in a certain ancestor because cellulases have already evolved through multiple clans into a large group of enzymes[39-42]including endocellulase (EC 3.2.1.4),exoglucanases (EC 3.2.1.74 and EC 3.2.1.91) and β-glucosidases (EC 3.2.1.21)[43-44].
These are important questions raised for the development of highly-productive cellulases in future efficiently and economically.Yet,cellulases have some specific characteristics in archaea,eukaryota,and bacteria[45],thus any abovementioned questions should be considered synergistically.To the best of our knowledge,no large-scale studies have been conducted to analyze the overall evolutionary relationship of cellulases so far,and yet there are no exhaustive studies that take pains to dig out detailed taxonomic lineage from superkingdom to organism for each cellulase for analysis.Hence,such studies would certainly provide valuable knowledge on our understanding of this important enzyme.In this study,2 416 cellulases,which were all available cellulases with clear and full taxonomic lineage from superkingdom to organism,in UniProtKB for release 2014_01 were analyzed to address the abovementioned questions[46].
1 Materials and methods
1.1Data
4 101 cellulase sequences were downloaded from UniProtKB,and this amount of cellulase sequences was all available cellulase sequences in UniProtKB for the release 2013_08 - Septempber 17,2013.Although these 4 101 cellulases were analyzed in our previous studies[47-48],their taxonomic lineages were verified against the UniProtKB for release 2014_01 - January 22,2014 for this study.This verification revealed that five entries (accession number,F7PZR3,L9JU04,D0BN91,L9KB35,L9KE38) had been deleted,so 4 096 cellulases remain,of which there were 736 and 3 360 cellulases with and without annotation of fragment.However,it did not mean that 3 360 cellulases without annotation of fragment were full-length sequences because the length for the 4 096 cellulases was ranged from 12 to 2 319 amino acids (414±211,mean±SD),the length for 736 cellulases with annotation of fragment was ranged from 12 to 1 232 amino acids (236±142,mean±SD),and the length for 3 360 cellulases without annotation of fragment was ranged from 34 to 2 319 amino acids (453±202,mean±SD).Clearly 3 360 cellulases without annotation of fragment should include fragmented cellulases.Of the 4 096 cellulases,2 came from viruses without taxonomic classifications and 239 cellulases were defined as unclassified sequences.Thus,3 855 cellulases moved towards to the next stage of analysis.
In order to accurately,precisely and reliably explore the evolutionary relationship of cellulases,only the cellulases that had full taxonomic classification from superkingdom,to kingdom,to phylum,to class,to order,to family,to genus,and to organism were included in statistical and phylogenetic analysis in this study.Accordingly,116 out of 133 archaea cellulases,1 744 out of 2 794 bacteria cellulases,and 556 out of 928 eukaryota cellulases met this criterion.Therefore only 2 416 cellulases were used in this study (Table S1,Supplementary Materials).Their fully taxonomic classifications for archaea,eukaryota and bacteria were listed in Tables S2,S3,and S4 in Supplementary Materials,respectively.
1.2Statistical analysis
This amount of data allows us to conduct a detailed statistical analysis to provide additional information on cellulases’ evolution.Mega software[49]was used to compute the average pairwise p-distance in each kingdom,phylum,class,order,family and genus.One-way ANOVA was used to compute statistical difference among clans.The model Ⅱ ANOVA was used to calculate inter-phyla,classes,orders,families and genera variance of the p-distance,and intra-phylum,class,order,family and genus variance,that is,the variance between groups and the variance within group[25-32].
1.3Phylogenetic analysis
The sequence alignment was conducted using Blast,Mega,and ClustalX[50],appropriately.The phylogenetic tree was constructed using ClustalX with neighbor-joining method,and presented using NJPlot[51].The phylogenetic trees were validated using 1 000 bootstrap replicates in ClustalX.
2 Results and discussion
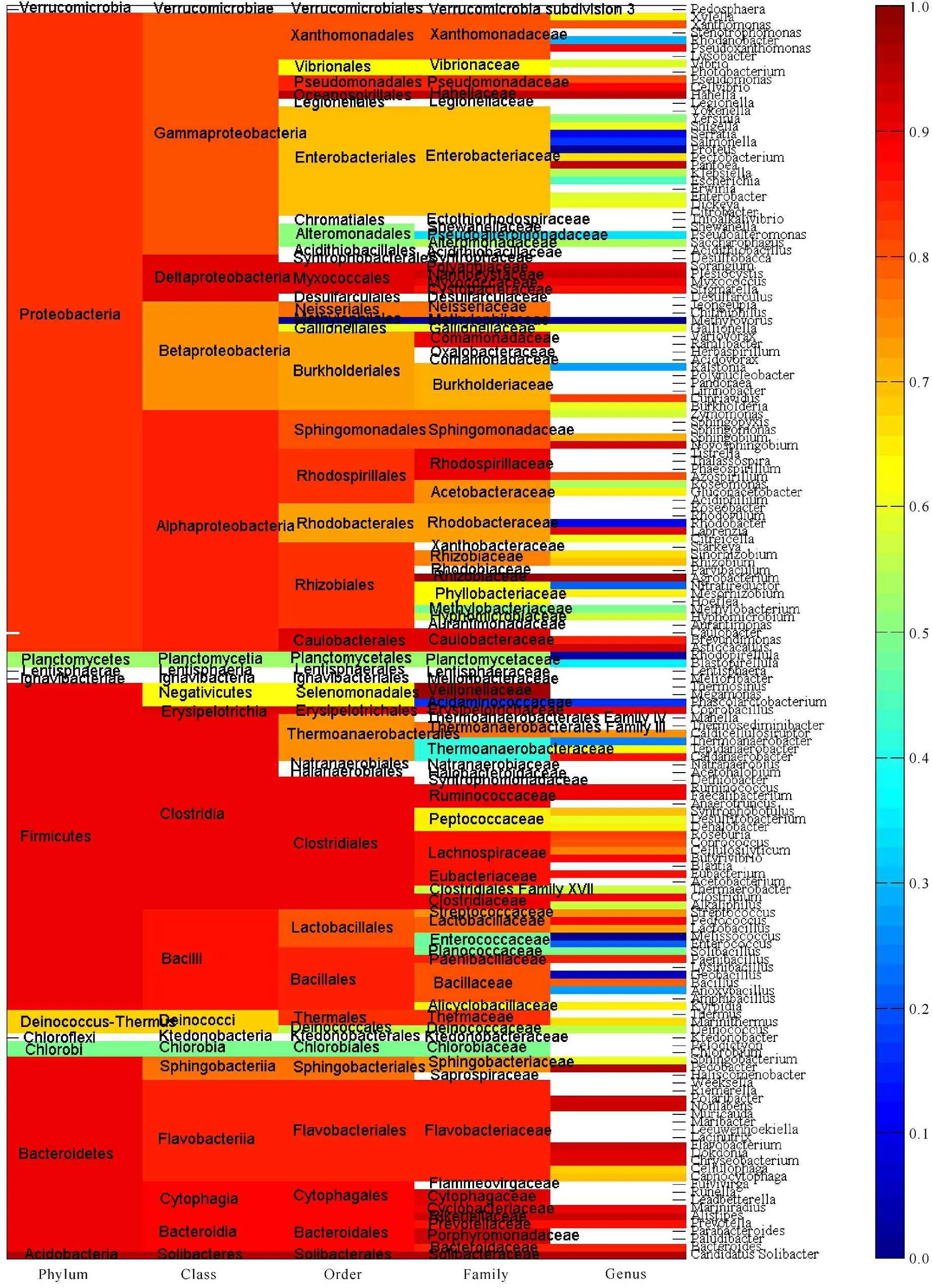
This large-scale statistical and phylogenitic study included 116,556 and 1 744 cellulases from archaea,eukaryota and bacteria,respectively.Thus they were treated separately and synergistically in the study.Figures 1-3 showed the average pairwise p-distance in archaea,eukaryota and bacteria cellulases,respectively.As the pairwise p-distance is the difference between two cellulase sequences in this context,so the average pairwise p-distance in a clan is the mean value of pairwise p-distance for each cellulase versus the rest of cellulases in the given clan.For example,Figure 1 showed that the average pairwise p-distance for each of 36 cellulases from phylum Crenarchaeota is 0.676 7,which was the mean value of pairwise p-distance for each of 36 cellulases versus the rest 35 cellulases and demonstrated the divergence of cellulases in Crenarchaeota.Obviously,it was not possible to conduct such analysis without the full knowledge on the taxonomic lineage of given cellulases.For instance,the average pairwise p-distance was obtained from 36 cellulase sequences in Crenarchaeota whereas the average pairwise p-distance was obtained from 80 cellulase sequences in phylum Euryarchaeota. The values of average pairwise p-distance varied greatly between clans,for example,the average pairwise p-distance was statistically larger in Crenarchaeota than in Euryarchaeota (0.676 7±0.076 9,n=36 vs 0.591 5±0.088 4,n=80,mean±SD,p<0.001).These very different average pairwise p-distances in different clans indicated different diversities in these cellulases.For another instance,the cellulases in order Sulfolobales were more diverse than that in order Methanococcales because the average pairwise p-distance was 0.627 8±0.047 3 for Sulfolobales (n=13) but was 0.224 4±0.026 0 for Methanococcales (n=14) as can be seen in Figure 1.The difference between Sulfolobales and Methanococcales could be attributed to various factors including random factors,such as selection of samples.Similar analysis can be applied to Figure 2 and Figure 3,which were far more reddish than Figure 1,that is,the clans in eukaryota and bacteria were far more diverse than the clans in archaea.Nevertheless,one-way ANOVA statistical test provided statistical difference among taxonomic classes,orders,families and genera.
The more cellulases are included in a study the more precisely evolutionary relationship can be found.Thus this study used all available amino acid sequences with full taxonomic lineage,which directly reflects the overall evolutionary relationship at genetic level.In view of Figures 1-3,the larger the average pairwise p-distance was,the more reddish the clan was.The difference in average pairwise p-distance suggested different evolutionary histories.For example,a faster evolution of cellulases in Sulfolobales than in Methanococcales,and an earlier expansion of cellulases in Sulfolobales than in Methanococcale presented in Figure 1.Hence,the ranking of average pairwise p-distance gave an estimate on cellulases’ evolution in each clan.On the other hand,the average pairwise p-distance had important experimental implication,and that is the efforts of genetic manipulation of cellulase should be directed to the clan whose average pairwise p-distance was large because mutants in this clan had a large chance to survive with desired enzymatic activities.Really,this could be the underlying reason why genetic manipulation was mainly conducted in eukaryota and bacteria and why mutants in eukaryota and bacteria accounted for a large portion of mutants.Typically,the bacteria,which lived in a more dynamic environment and whose DNA flows were varying,were more diverse in their evolution[52].
Actually,the average pairwise p-distance was quite large in some cases,even for cellulases from the same species.For example,two cellulases,A8MDC6 and A8MAL9,came from Archaea,Crenarchaeota,Thermoprotei,Thermoproteales,Thermoproteaceae,Caldivirga,Caldivirga maquilingensis,strain ATCC 700844/DSM 13496/JCM 10307/IC-167,but their p-distance was quite large (0.646 6),because 64.66% amino acids were different between them.However,this was because the strain gene produces two cellulases at two different sites.Clearly,this was not a simple duplication of cellulase gene,but was more likely to acquire the gene from a sister clan.
Different patterns in Figures 1-3 explained different speeds in the evolution of cellulases in different kingdoms.In reality,the evolution of cellulases should be more complicated than any man-made analysis because historical data indicated that there were 14 000 fungal species that were able to degrade cellulose[53],which easily overwhelmed the number of cellulases documented in any databases.Often,dramatic changes in genomes marked particular evolutionary events,which were due to the gain and loss of genes,and were found in alpha-proteobacteria[54]and betaproteobacteria[55],which also produced cellulases.And Firmicutes,which produced cellulases too,could acquire genes for photosynthetic apparatus[56].
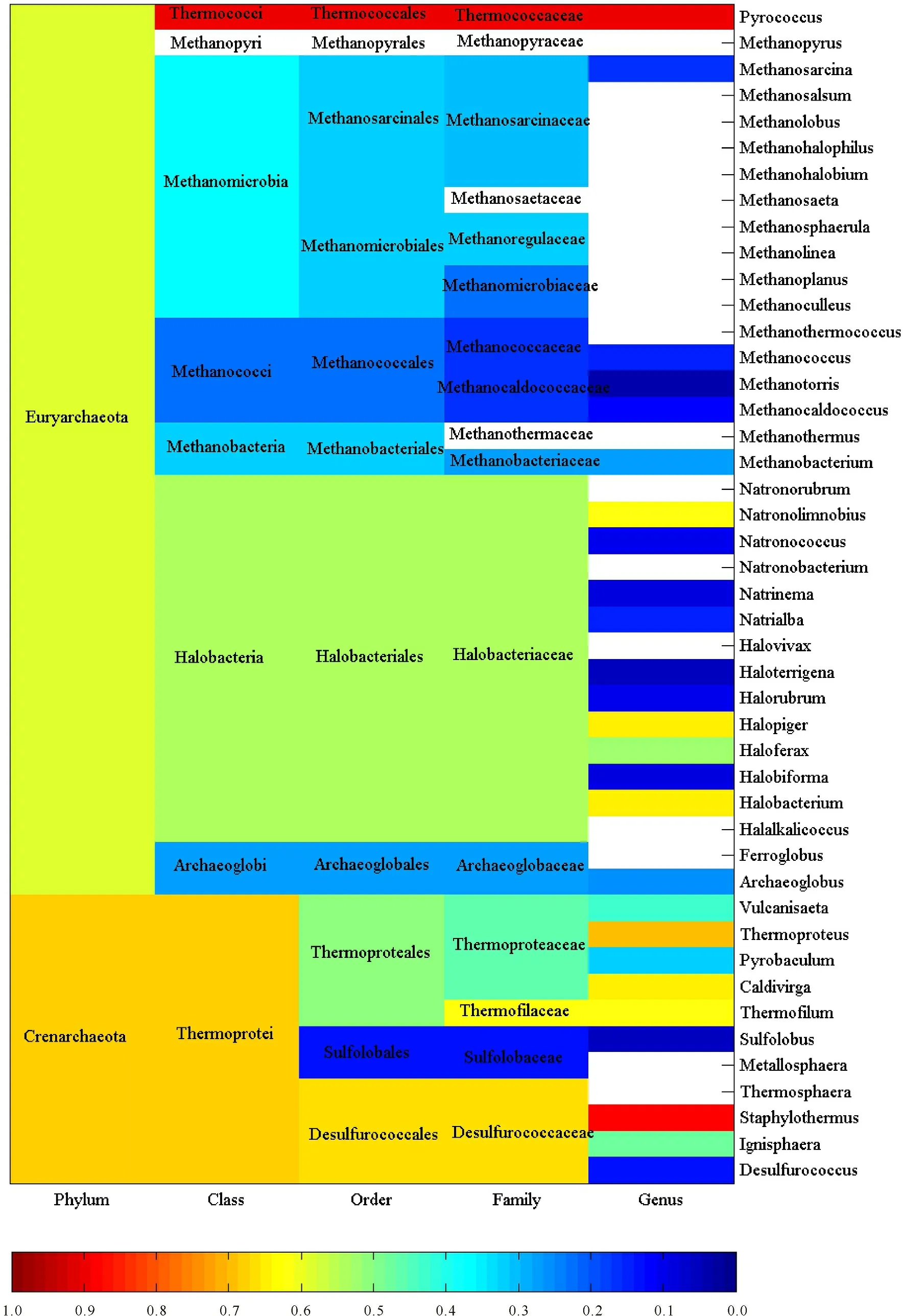

Blank:Average pairwise p-distance is unavailable
Blank: Average pairwise p-distance is unavailable
Blank:Average pairwise p-distance is unavailable
空白:平均成对p距离未获得
Fig.3Average pairwise p-distance of cellulases from bacteria
图3细菌纤维素酶的平均成对p距离
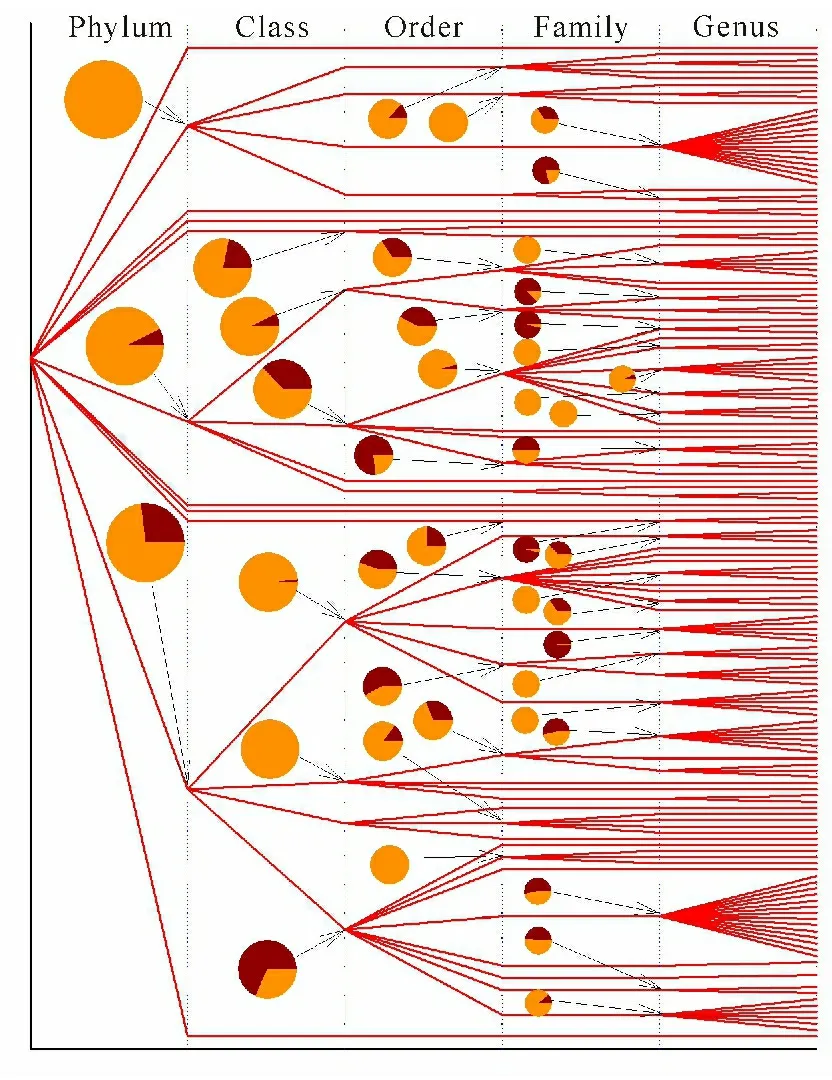
Figures 4-6 revealed inter-clan variance and intra-clan variance with respect to the average pairwise p-distance in archaea,eukaryota and bacteria,respectively,by means of model Ⅱ ANOVA.Again,archaea (Figure 4) could serve as an example,because the number of cellulases collected from archaea was the smallest among them.This type of figures could be read as follows: (i) each line indicated the route that a member of clan had so far evolved from kingdom to phylum,to class,to order,to family and finally to genus; and (ii) each pie indicated the inter-clan variance (dark color) and the intra-clan variance (bright color) with respect to the average pairwise p-distance.It turned out in Figure 4,for example,that the cellulases from archaea came from two phyla Crenarchaeota (upper line) and Euryarchaeota (lower line),while the data for Euryarchaeota allowed the computation of the inter-class variance (11.70%) and the intra-class variance (88.30%) as shown in the dark and bright portions in the pie.Following this,the cellulases from Thermoprotei (upper line at class level) were composed of three orders,Desulfurococcales,Sulfolobales and Thermoproteales,of which the inter-order and intra-order variances were 42.82% and 57.18%,and so on.Figure 5 got more data,so more interesting evolutionary line could be tracked.Basically,eukaryota evolved into three phyla,Fungi (upper line),Metazoa (middle line) and Viridiplantae (lower line).However,two of them revealed that their inter-phylum variance (dark portion in pies) was followed by a continuing increase in inter-clan variance leading to more divergence.It was interesting to note a sort of pattern in Figure 5,namely,large inter-clan variances appeared at a lower part of Figure 5.However,this pattern was not clearly visible in Figure 6,which showed the inter- and intra-clan variances of the p-distance in bacteria.
Using model Ⅱ ANOVA,the variance of average pairwise p-distance was further divided into intra- and inter-clan variances and displayed as bright and dark portions in a pie.First of all,this type of figures could clearly show the evolutionary pathway of each member in a clan,which was only accomplished by obtaining full taxonomic lineage from UniProtKB.Secondly,a line indicated at which level that a member in a clan had crossed a taxonomic boundary line and evolved into another clan.Thirdly,a clan could embrace a certain number of members or just a single member.Fourthly,a pie not only recorded what happened in the past but also implicated what might happen in the future.What their implications were when the dark portion in a pie was smaller than the bright portion,the inter-clan variance of the p-distance was smaller than the intra-clan variance,and the cellulases in this clan was more likely to evolve along a taxonomic line and limit their evolution within taxonomic boundary lines.Thus,their genetic materials were inherited vertically.By contrast,when the dark portion in a pie was larger than the bright portion,the inter-clan variance of the p-distance was larger than the intra-clan variance,and the cellulases in the clan were more likely to evolve across a taxonomic line and would not limit their evolution within taxonomic boundary lines.Thus,their genetic materials were likely to experience horizontal gene transfer and develop multiple clan evolution[57].In this manner,genetic manipulation could be set in line with model Ⅱ ANOVA analysis.For example,a gene from a certain genus with desired qualification could be cloned and expressed in the species whose intra-genus variance was large if the gene came from the same genus,otherwise it could be cloned and expressed in the species whose inter-genus variance was small if the gene came from a different genus in order to increase the successful rate.Also,one of the most important things to realize was that the inter-clan variance could be connected with the horizontal transfer of gene[36-38,58-59].
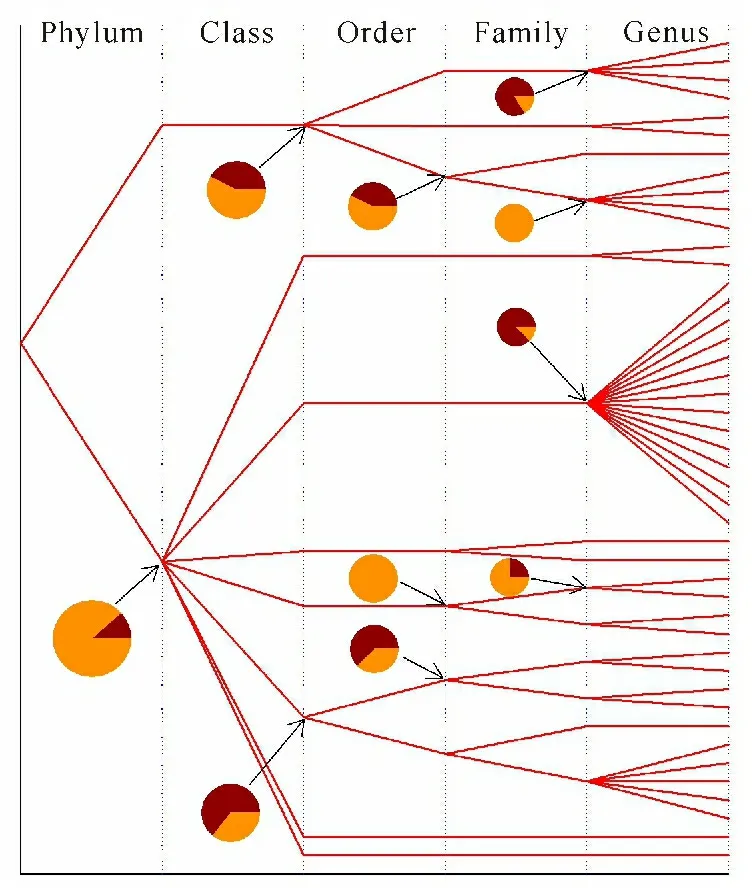
The bifurcation is the point across taxonomic boundary;Pies show the inter-clan variance (dark color) and intra-clan variance (bright color);Taxonomic names can be found in Supplementary Material Table S5
分叉代表跨种属的分界点;饼图表示种属间差异(红褐色)和种属内差异(橙黄色);各种属名称见补充材料表S5
Fig.4Comparison between intra-clan and inter-clan variances from archaea cellulases
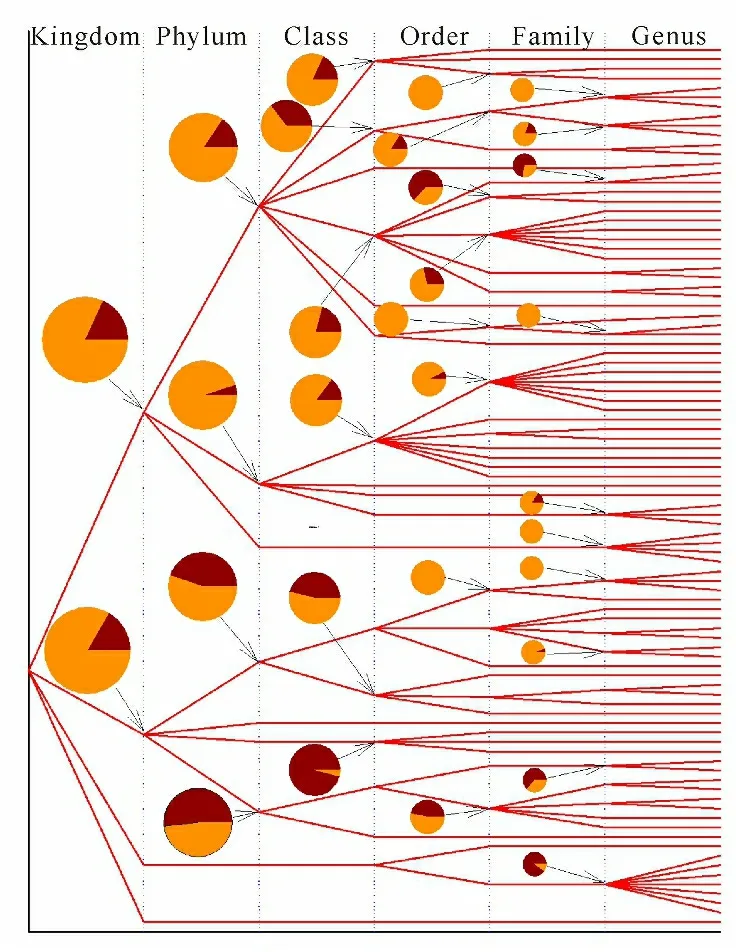
图4 古菌纤维素酶的种属内差异和种属间差异
The bifurcation is the point across taxonomic boundary;Pies show the inter-clan variance (dark color) and intra-clan variance (bright color);Taxonomic names can be found in Supplementary Material Table S6
分叉代表跨种属的分界点;饼图表示种属间差异(红褐色)和种属内差异(橙黄色);各种属名称见补充材料表S6
Fig.5Comparison between intra-clan and inter-clan variances from eukaryota cellulases
图5真核生物纤维素酶的种属内差异和种属间差异
The bifurcation is the point across taxonomic boundary;Pies show the inter-clan variance (dark color) and intra-clan variance (bright color);Taxonomic names can be found in Supplementary Material Table S7
分叉代表跨种属的分界点;饼图表示种属间差异(红褐色)和种属内差异(橙黄色);各种属名称见补充材料表S7
Fig.6Comparison between intra-clan and inter-clan variances from bacterial cellulases
图6细菌纤维素酶的种属内差异和种属间差异
Figures 7 and 8 displayed phylogenetic analyses of cellulases from archaea and eukaryota,respectively.As can be seen in Figure 7,the cellulases from aechaea evolved into two branches similar to what had been arranged according to taxonomic lineage in Figure 4.The most upper branch in Figure 7 included the cellulases F4G2P2,Q97VS7,Q7LWG2,C3MKY0,D2PFG3,C3N9U6,C4KL23,C3N1U7,C3MUI4,C3NM45,F0NJB9,M9UC91 and F0NE90.Besides the cellulase F4G2P2,all the rest belonged to the genus Sulfolobus,which was in good agreement with taxonomic lineage.However,the cellulase F4G2P2 belonged to the genus Metallosphaera,whose bifurcation should be at family level,Sulfolobaceae.Accordingly,the most upper line in Figure 7 suggested the cellulase F4G2P2 evolved at a very earlier stage of evolution,which was different from taxonomic lineage in Figure 4.In addition,the phylogenetic tree in Figure 7 revealed a faster speed of evolution in certain clusters.For example,the cellulases A6VG61 and A9AAJ0 experienced 12 evolutionary steps,and eventually belonged to Methanococcus.On the other hand,both cellulases F4G2P2 and M0GK59 experienced a slower speed of evolution because they had only two evolutionary steps,and eventually belonged to Metallosphaera and Haloferax.These were very suggestive because evolutionary pathways of Methanococcus,Metallosphaera and Haloferax might give some clue on the underlying mechanism for different speeds of evolution,which were subject to the abundant cellulose as the selective pressure[60],and these results were supported by high bootstrap values.

Fig.7Phylogenetic tree of cellulases from archaea
图7古菌纤维素酶的系统进化树
Figure 8 demonstrated just a portion of phylogenetic tree composed of 203 cellulases from eukaryota because of limit of space.As a matter of fact,the whole picture of phylogenetic tree in Figure 5 gave an impression that there were two clusters.The upper cluster (light green bar) aggregated 85 cellulases under accession numbers of I6U--- as well as I6V--- that belonged to three genera,Panicum,Setaria and Urochloa,and cellulase D5K8E7 from the genus Leptographium.And the bottom cluster (dark green bar) aggregated 34 cellulases under accession numbers of E9NH-- that exclusively belong to the genus Pristionchus.Very interestingly,12 cellulases under accession numbers of F1CZ-- (red colored cluster in Figure 8) also belonged to Pristionchus,but their locations were far away from those 34 cellulases under accession numbers of E9NH-- (bottom cluster in Figure 8).Consequently the cellulase clans E9NH-- and F1CZ-- followed completely different evolutionary pathways because a chunk of 68 cellulases from various species,which account for 12.2% of 557 species from eukaryota,evolved between them.
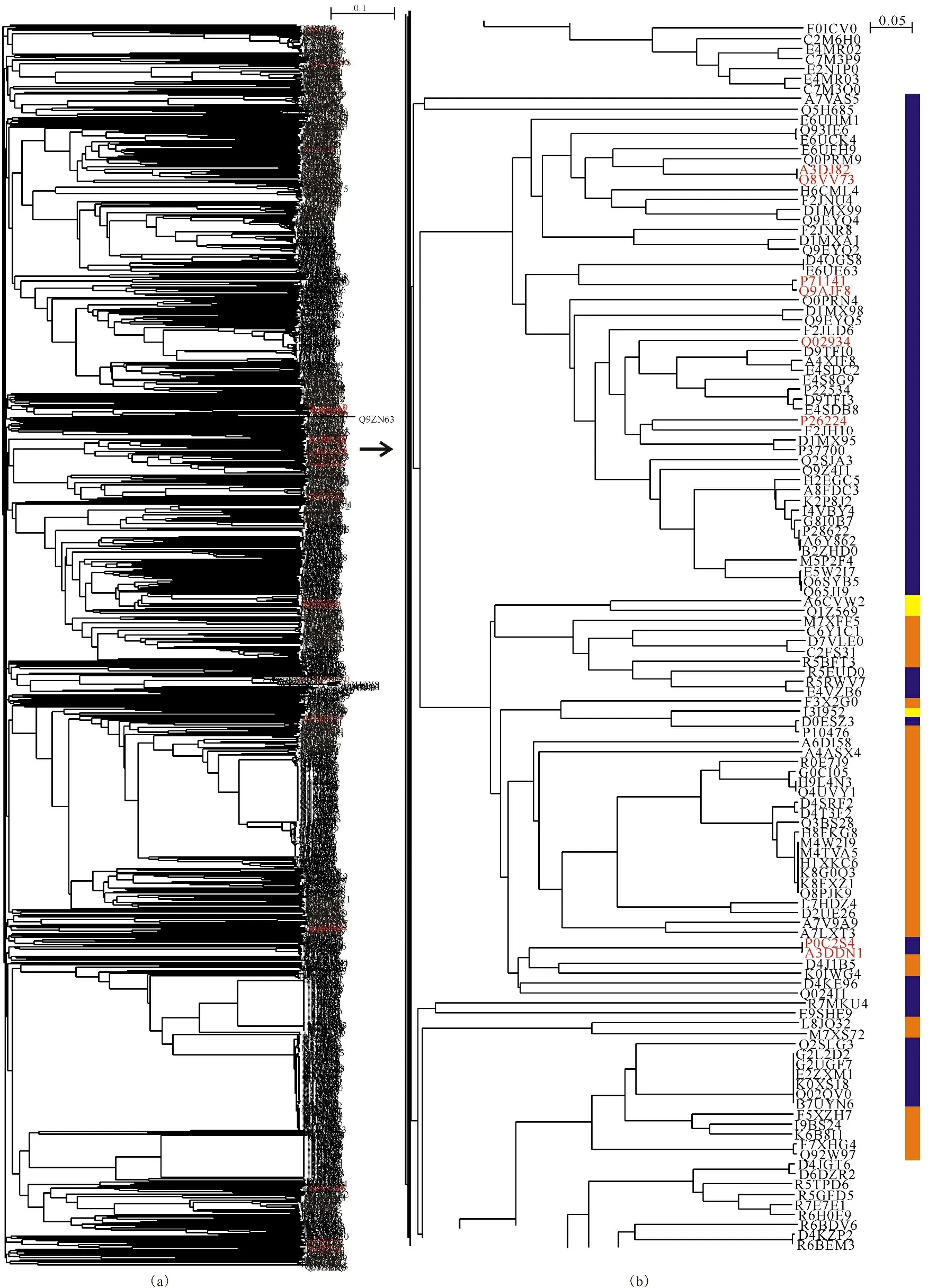
Figure 9a displayed the full phylogenetic tree of 1 744 cellulases from bacteria,where the cellulases from Clostridium thermocellum were marked with red,which distributed from top to bottom with their accession numbers P15329,P0C2S5,A3DH67,P10477,P0C2S3,A3DJ77,P0C2S2,A3DJ82,Q8VV73,P71141,Q9AJF8,Q02934,P26224,P0C2S4,A3DDN1,E6USM0,A3DD14,D1NI26,C7HDK9,Q46391,Q9R670,A3DC29,P16218,P71140,P04956,Q05332,P0C2S1,A3DCJ4.Figure 9b presented a portion of phylogenetic tree of these cellulases with respect to their characteristics of aerobic,facultative and anaerobic.In general,bacteria were different in their ability to deal with cellulose with respect to whether a bacterium was aerobic or anaerobic[61].For instance,Clostridium thermocellum is anaerobic[62-63],whose cellulases distributed in the liquid phase and on the cell surface,and produced also a spectrum of cellulolytic and hemicellulolytic enzymes in a multienzyme form known as cellulosome.From Figure 9b,a pattern revealed obvious that anaerobic and aerobic species were separated by facultative species (yellow bar).Thus,it was very informative and important to realize that such evolutionary pattern was in consistence with the metabolism property of species,suggesting that they had evolved together from a common ancestor[18],and then evolved to aerobic,facultative and anaerobic to adapt to different living conditions in multiple times.Furthermore,this pattern goes as Darwin put: “The characters do not make the genus,but the genus gives the characters.” Indeed,the mixture of these bacteria of distantly related taxonomic clans implicated candidate gene that transmitted to adapt to aerobic or anaerobic lifestyles[64].
When overall evolution of cellulases was analyzed from all species of archaea,eukaryota and bacteria,so many issues could be addressed and many interesting things could be dug out,but here the cellulases from archaea were discussed for conciseness because of limit of space.In the full phylogenetic tree of 2 416 cellulases (Figure 10a),the cellulases from archaea were marked as red and distributed in 5 clusters (Figure 10b-10f).In general,the cellulases from archaea evolved close with those from bacteria,indicating that the cellulases from archaea might be more likely to be evolved from bacteria.

Light green bar,the cellulases under accession numbers of I6U--- or I6V---;Dark green bar,the cellulases under accession numbers of E9NH--;The red colored cluster,the cellulases under accession numbers of F1CZ--
浅绿柱,I6U---或I6V---纤维素酶;深绿柱,E9NH--纤维素酶;红色簇,F1CZ--纤维素酶
Full phylogenetic tree of 1 744 cellulases from bacteria (a) with the cellulases of Clostridium thermocellum marked with red,and a part phylogenetic tree (b) of them (arrow);The characteristics of aerobic,facultative and anaerobic are indicated by orange,yellow and purple bars,respectively
1 744个细菌纤维素酶的完整进化树(a);其中来自Clostridium thermocellum的纤维素酶以红色表示,箭头引出其部分进化树(b);好氧、兼氧和厌氧的细菌分别以橙色、黄色和深蓝色表示

Fig.9 Phylogenetic tree from bacteria
Phylogenetic tree of all 2 416 cellulases (a),where the cellulases from archaea were marked with red,and their distribution with a part of phylogenetic tree in (b) to (f).The cellulases from archaea,eukaryota and bacteria are indicated by pink,green and blue bars,respectively
2 416个纤维素酶的完整进化树(a);其中古菌纤维素酶标记为红色,(b)~(f)表示在进化树中的分布。古菌、真核生物和细菌的纤维素酶分别用粉色、绿色和蓝色表示
Fig.10Phylogenetic tree from archaea,eukaryota and bacteria
图10古菌、真核生物和细菌的系统发育树
The cellulases from the same clan would have a large chance to cluster together,while a single outlier from a cluster could be the result of horizontal transfer of genetic materials across species[61].It is interesting to find that some cellulases distributed in the phylogenetic tree to interweave with clans,and some examples were a cellulase from archaea A1RW32 distributing among a cluster of bacteria (Figure 10f),two cellulases from eukaryota R4V447 and H1W3B9 distributing in the cluster of cellulases from bacteria (Figure 10b),a cellulase from bacteria Q53652 distributing among the cellulases of archaea (Figure 10c),and so on.Horizontal gene transfer had played an important role in biological evolution,and contributed the vast numbers of organisms[36-38,59,61,65],which could benefit from new gene packages to change their diet,to colonize new habitats,or broaden survive conditions[66].Although the cellulase evolution in Figure 10 appeared to undergo a series of developments,cellulase certainly appeared more later than the appearance of cellulose,which was associated with the development of algae and land plants.Therefore selective pressure should be related to cellulose that was available to cellulase leading to different evolutionary rates[52]as witnessed by long branch subtending in Figures 7-10,suggesting positive selection[67].
In this study,the average pairwise p-distance for each clan,and inter- and intra-clan variances provide additional information on the overall evolutionary relationship of cellulases because this additional information can play a complementary role for phylogenetic trees,to which different views were recently given[68-69].Also,it is because the biological evolution ranges from genes to communities[70],which demands new tools and methods.In this context,the inter-clan variance might serve as a numeric measure to estimate the lateral gene transfer[71]and gene loss.
Technical issues might be worthy mentioning in order to enlighten such large-scale studies in future.The exponential increase in the number of completely sequenced species challenges computational power radically,for example,a recent study on life tree contains 1 133 organisms[72].It is always a choice between protein and DNA in phylogenetic analysis,while cellulase protein sequences were used in this study because such a quantity of sequences makes it difficult to run alignment using DNA.Indeed,Clustal Omega and MUSCLE used in European Bioinformatics Institute limit 2 000 and 500 sequences for alignment so it is recommended to align translated proteins when sequence data containing codons and alignment at amino acid sequence level are generally more reliable than that at the nucleotide level because amino acid sequences evolve much more slowly[73].Also,it is hard to construct an artistic phylogenetic tree in such a large-scale phylogenetic analysis.For example,phylogenetic tree visualization software TreeView can only present 1 000 members.Nevertheless,many important issues can be dug out from the phylogenetic trees in those figures,especially in Figure 10.Due to the limit of space,these issues would not be addressed over here,however these analysis doubtless would benefit to our knowledge on the cellulases’ revolution.
3 Conclusions
This study conducted large-scale statistical and phylogenetic analyses of amino acid sequences of cellulases,which provides new insights on the overall evolutionary relationship of cellulases from archaea,bacteria and eukaryota.The difference in average pairwise p-distance suggested different evolutionary histories,and the variance of average pairwise p-distance was further divided into inter-clan variance and intra-clan variance using model Ⅱ ANOVA.The cellulases with larger intra-clan variance were more likely to evolve within taxonomic boundary lines,and their genetic materials were inherited vertically.On the contrary,the cellulases with larger inter-clan variance were more likely to evolve across a taxonomic boundary line,and their genetic materials were likely to experience horizontal gene transfer.Therefore,the partition of average pairwise p-distance into inter-clan variance and intra-clan variance provides numeric measures,which can be useful to estimate the lateral gene transfer and gene loss.As additional information on the overall evolutionary relationship of cellulases,they can play a complementary role for understanding the phylogenetic trees,where most cellulases underwent different paths and bifurcations,and formed intertwined clusters.
Supplementary Materials:
Table S1 Accession numbers for 2 416 used in this study
Table S2 Taxonomic classification of 116 cellulases from archaea used in this study
Table S3 Taxonomic classification of 556 cellulases from eukaryota used in this study
Table S4 Taxonomic classification of 1 796 cellulases from bacteria used in this study
Table S5 Taxonomic names for Figure 4
Table S6 Taxonomic names for Figure 5
Table S7 Taxonomic names for Figure 6
N.B.:Supplementary Materials can be available from the authors.
[1]PHITSUWAN P,LAOHAKUNJIT N,KERDCHOECHUEN O,et al.Present and potential applications of cellulases in agriculture,biotechnology,and bioenergy[J].Folia Microbiol,2013,58(2):163-176.
[2]HARRIS P V,XU F,KREEL N E,et al.New enzyme insights drive advances in commercial ethanol production[J].Curr Opin Chem Biol,2014,19:162-170.DOI:10.1016/j.cbpa.2014.02.015.
Trichoderma atroviride mutants with enhanced production of cellulase and β-glucosidase on pretreated willow[J].Enzyme Microb Technol,2008,43(1):48-55.
[4]AHAMED A,VERMETTE P.Enhanced enzyme production from mixed cultures of Trichoderma reesei RUT-C30 and Aspergillus niger LMA grown as fed batch in a stirred tank bioreactor[J].Biochem Eng J,2008,42(1):41-46.
[5]WANG M Y,ZHAO Q S,YANG J H,et al.A mitoge-
n-activated protein kinase Tmk3 participates in high osmolarity resistance,cell wall integrity maintenance and cellulase production regulation in Trichoderma reesei[J].PLoS One,2013,8(8):e72189.
[6]QIN Y Q,WEI X M,SONG X,et al.Engineering endoglucanase Ⅱ from Trichoderma reesei to improve the catalytic efficiency at a higher pH optimum[J].J Biotechnol,2008,135(2):190-195.
[7]FENEL F,LEISOLA M,JNIS J,et al.A de novo designed N-terminal disulphide bridge stabilizes the Trichoderma reesei endo-1,4-β-xylanase Ⅱ[J].J Biotechnol,2004,108(2):137-143.
[8]TRUDEAU D L,LEE T M,ARNOLD F H.Engineered thermostable fungal cellulases exhibit efficient synergistic cellulose hydrolysis at elevated temperatures[J].Biotechnol Bioeng,2014,111:2390-2397.
[9]CHEN M,KOSTYLEV M,BOMBLE Y J,et al.Experimental and modeling studies of an unusual water-filled pore structure with possible mechanistic implications in family 48 cellulases[J].J Phys Chem B,2014,118:2306-2315.
[10]CHEN L Q,DRAKE M R,RESCH M G,et al.Specificity of O-glycosylation in enhancing the stability and cellulose binding affinity of Family 1 carbohydrate-binding modules[J].Proc Natl Acad Sci USA,2014,111:7612-7617.
[11]FURUKAWA K,ICHIKAWA S,NIGORIKAWA
M,et al.Enhanced production of reducing sugars from transgenic rice expressing exo-glucanase under the control of a senescence-inducible promoter[J].Transgenic Res,2014,23:531-537.
[12]GARDNER R G,WELLS J E,RUSSELL J B,et al.The cellular location of Prevotella ruminicola β-1,4-D-endoglucanase and its occurrence in other strains of ruminal bacteria[J].Appl Environ Microbiol,1995,61:3288-3292.
[13]THOMAS L,JOSEPH A,GOTTUMUKKALA L
D.Xylanase and cellulase systems of Clostridium sp.:An insight on molecular approaches for strain improvement[J].Bioresour Technol,2014,158:343-350.
[14]ALVES P D D,DE FARIA SIQUEIRA F,FACCHIN S,et al.Survey of microbial enzymes in soil,water,and plant microenvironments[J].Open Microbiol J,2014,8:25-31.
[15]WATANABE H,TOKUDA G.Animal cellulases[J].Cell Mol Life Sci,2001,58:1167-1178.
[16]LO N,WATANABE H,SUGIMURA M.Evidence
for the presence of a cellulase gene in the last common ancestor of bilaterian animals[J].Proc Biol Sci,2003,270 (Suppl 1):S69-S72.
[17]DASHTBAN M,SCHRAFT H,QIN W S.Fungal
bioconversion of lignocellulosic residues: Opportunities & perspectives[J].Int J Biol Sci,2009,5:578-595.
[18]DARWIN C.The Origin of Species:By Means of Natural Selection,or The Preservation of Favoured Races in the Struggle for Life[M].6th Edition.London: John Murray,1876.
[19]YAN Y T,SMANT G,STOKKERMANS J,et al.
Genomic organization of four β-1,4-endoglucanase genes in plant-parasitic cyst nematodes and its evolutionary implications[J].Gene,1998,220:61-70.
[20]SUKHARNIKOV L O,ALAHUHTA M,BRUNEC-
KY R,et al.Sequence,structure,and evolution of cellulases in glycoside hydrolase family 48[J].J Biol Chem,2012,287:41068-41077.
[21]KORPOLE S,SHARMA R,VERMA D.Characterization and phylogenetic diversity of carboxymethyl cellulase producing Bacillus species from a landfill ecosystem[J].Indian J Microbiol,2011,51:531-535.
[22]PEI J J,PANG Q,ZHAO L G,et al.Thermoanaer-
obacterium thermosaccharolyticum β-glucosidase:A glucose-tolerant enzyme with high specific activity for cellobiose[J].Biotechol Biofuels,2012,5:31.
[23]NEI M,KUMAR S.Molecular Evolution and Phylogenetics[M].Oxford: Oxford University Press,2000.
[24]GLADDEN J M,PARK J I,BERGMANN J,et al.
Discovery and characterization of ionic liquid-tolerant thermophilic cellulases from a switchgrass-adapted microbial community[J].Biotechnol Biofuels,2014,7:15.
[25]SOKAL R R,ROHLF F J.Biometry:The Principles and Practices of Statistics in Biological Research[M].3rd edition.New York:W.H.Freeman,1995:203-218.
[26]WU G,BARALDO M,FURLANUT M.Inter-patient and intra-patient variations in the baseline tapping test in patients with Parkinson’s disease[J].Acta Neurol Belg,1999,99:182-184.
[27]YAN S,WU G.Rationale for cross-species infection
and cross-subtype mutation in hemagglutinins from influenza A virus[J].Interdiscip Sci Comput Life Sci,2009,1:303-307.
[28]YAN S,WU G.Evidence obtained from ANOVA to reason cross-species infection and cross-subtype mutation in neuraminidases of influenza A viruses[J].Transbound Emerg Dis,2010,57:254-261.
[29]YAN S,WU G.Evidence for cross-species infection and cross-subtype mutation in influenza A matrix proteins[J].Viral Immunol,2010,23:105-111.
[30]YAN S,WU G.Possible reason for cross-species and cross-subtype reassortment in polymerase basic protein 2 from influenza A virus[J].Protein Pept Lett,2011,18:434-439.
[31]YAN S,WU G.Small variations between species/
subtypes attributed to reassortment evidenced from polymerase basic protein 1 with other seven proteins from influenza A virus[J].Transbound Emerg Dis,2013,60(2):110-119.
[32]YAN S,WU G.Possibility of cross-species/subtype
reassortments in influenza A viruses: An analysis on nonstructural protein variations[J].Virulence,2013,4:716-725.
[33]SCHLIEP K,LOPEZ P,LAPOINTE F J,et al.Harvesting evolutionary signals in a forest of prokaryotic gene trees[J].Mol Biol Evol,2011,28:1393-1405.
[34]MCINERNEY J O,COTTON J A,PISANI D.The prokaryotic tree of life: Past,present… and future?[J].Trends Ecol Evol,2008,23(5):276-281.
[35]O’HARA R J.Population thinking and tree thinking in systematics[J].Zool Scripta,1997,26:323-329.
[36]SMANT G,STOKKERMANS J P,YAN Y,et al.Endogenous cellulases in animals:Isolation of β-1,4-endoglucanase genes from two species of plant-parasitic cyst nematodes[J].Proc Natl Acad Sci USA,1998,95:4906-4911.
[37]SCHOLL E H,BIRD D M.Computational and phylogenetic validation of nematode horizontal gene transfer[J].BMC Biol,2011,9:9.
[38]MAYER W E,SCHUSTER L N,BARTELMES G,et al.Horizontal gene transfer of microbial cellulases into nematode genomes is associated with functional assimilation and gene turnover[J].BMC Evol Biol,2011,11:13.
[39]BARTHELMES J,EBELING C,CHANG A,et al.BRENDA,AMENDA and FRENDA: The enzyme information system in 2007[J].Nucleic Acids Res,2007,35:D511-D514.
[40]CHANG A,SCHEER M,GROTE A,et al.BRENDA,AMENDA and FRENDA the enzyme information system: New content and tools in 2009[J].Nucleic Acids Res,2009,37:D588-D592.
[41]SCHEER M,GROTE A,CHANG A,et al.BRENDA,the enzyme information system in 2011[J].Nucleic Acids Res,2011,39:D670-D676.
[42]SCHOMBURG I,CHANG A,PLACZEK S,et al.
BRENDA in 2013: Integrated reactions,kinetic data,enzyme function data,improved disease classification:New options and contents in BRENDA[J].Nucleic Acids Res,2013,41:D764-D772.
[43]WATANABE H,TOKUDA G.Cellulolytic systems
in insects[J].Ann Rev Entomol,2010,55:609-632.
[44]ZVERLOV V V,SCHANTZ N,SCHWARZ W H.A major new component in the cellulosome of Clostridium thermocellum is a processive endo-beta-1,4-glucanase producing cellotetraose[J].FEMS Microbiol Lett,2005,249:353-358.
[45]LYND L R,WEIMER P J,VAN ZYL W H,et al.Microbial cellulose utilization:Fundamentals and biotechnology[J].Microbiol Mol Biol Rev,2002,66:506-577.
[46]The UniProt Consortium.The universal protein reso-
urce (UniProt) in 2010[J].Nucleic Acids Res,2010,38:D142-D148.
[47]YAN S,WU G.Secretory pathway of cellulase:A
mini-review[J].Biotechnol Biofuels,2013,6:177.
[48]YAN S,WU G.Signal peptide of cellulase[J].Appl
Microbiol Biotechnol,2014,98:5329-5362.
[49]TAMURA K,STECHER G,PETERSON D,et al.
MEGA6:Molecular evolutionary genetics analysis version 6.0[J].Mol Biol Evol,2013,30(12):2725-2729.
[50]LARKIN M A,BLACKSHIELDS G,BROWN N P,et al.Clustal W and Clustal X version 2.0[J].Bioinformatics,2007,23:2947-2948.
[52]WILMES P,SIMMONS S L,Denef V J,et al.The dynamic genetic repertoire of microbial communities[J].FEMS Microbiol Rev,2009,33:109-132.
[53]MANDELS M,STERNBERG D.Recent advances in
cellulase technology[J].J Ferment Technol,1976,54:267-286.
[54]BLANC G,OGATA H,ROBERT C,et al.Reductive genome evolution from the mother of Rickettsia[J].PLoS Genet,2007,3:e14.
[55]MORAN N A,MIRA A.The Process of genome shri-
nkage in the obligate symbiont Buchnera aphidicola[J].Genome Biol,2001,2(12):Research0054.1.DOI:10.1186/gb-2001-2-12-research0054.
[56]MEINEL T,KRAUSE A.Meta-analysis of general
bacterial subclades in whole-genome phylogenies using tree topology profiling[J].Evol Bioinform,2012,8:489-525.
[57]VON WITTGENSTEIN N J,LE C H,HAWKINS B J,et al.Evolutionary classification of ammonium,nitrate,and peptide transporters in land plants[J].BMC Evol Biol,2014,14:11.
[58]SCHOLL E H,THORNE J L,MCCARTER J P,et al.Horizontally transferred genes in plant-parasitic nematodes: A high-throughput genomic approach[J].Genome Biol,2003,4:R39.
[59]PLUCAIN J,HINDRÉ T,LE GAC M,et al.Epistasis and allele specificity in the emergence of a stable polymorphism in Escherichia coli[J].Science,2014,343:1366-1369.
[60]LIANG G M,JIANG X P.Positive selection drives
lactoferrin evolution in mammals[J].Genetica,2010,138:757-762.
[61]RAINEY F A,DONNISON A M,JANSSEN P H,et al.Description of Caldicellulosiruptor saccharolyticus gen.nov.,sp.nov: An obligately anaerobic,extremely thermophilic,cellulolytic bacterium[J].FEMS Microbiol Lett,1994,120:263-266.
[62]SCHWARZ W.The cellulosome and cellulose degradation by anaerobic bacteria[J].Appl Microbiol Biotechnol,2001,56:634-649.
[63]RAMAN B,PAN C L,HURST G B,et al.Impact of pretreated switchgrass and biomass carbohydrates on Clostridium thermocellum ATCC 27405 cellulosome composition: A quantitative proteomic analysis[J].PLoS One,2009,4:e5271.
[64]LEGAULT B A,LOPEZ-LOPEZ A,ALBA-CASADO J C,et al.Environmental genomics of “Haloquadratum walsbyi” in a saltern crystallizer indicates a large pool of accessory genes in an otherwise coherent species[J].BMC Genomics,2006,7:171.
[65]DARMON E,LEACH D R.Bacterial genome instability[J].Microbiol Mol Biol Rev,2014,78:1-39.
[66]MITREVA M,SMANT G,HELDER J.Role of horizontal gene transfer in the evolution of plant parasitism among nematodes[J].Methods Mol Biol,2009,532:517-535.
[67]TSANTES C,STEIPER M E.Age at first reproduction explains rate variation in the strepsirrhine molecular clock[J].Proc Natl Acad Sci USA,2009,106:18165-18170.
[68]DAGAN T,MARTIN W.Getting a better picture of microbial evolution en route to a network of genomes[J].Philos Trans R Soc Lond B Biol Sci,2009,364:2187-2196.
[69]RAGAN M A,BEIKO R G.Lateral genetic transfer: Open issues[J].Philos Trans R Soc Lond B Biol Sci,2009,364:2241-2251.
[70]BAPTESTE E,BOUCHER Y.Lateral gene transfer
challenges principles of microbial systematics[J].Trends Microbiol,2008,16:200-207.
[71]LO I,DENEF V J,VERBERKMOES N C,et al.
Strain-resolved community proteomics reveals recombining genomes of acidophilic bacteria[J].Nature,2007,446:537-541.
[72]POWELL S,SZKLARCZYK D,TRACHANA K,et
al.eggNOG v3.0: Orthologous groups covering 1133 organisms at 41 different taxonomic ranges[J].Nucleic Acids Res,2012,40(D1):D284-D289.
[73]Molecular Evolutionary Genetic Analysis.Manual of
MEGA Software[CP].1993-2009:60.
(责任编辑:米慧芝)
2015-12-13
严少敏(1958-),女,研究员,主要从事计算变异学和生物信息学研究。
古菌、细菌和真核生物纤维素酶的整体进化关系
YAN Shaomin,WU Guang**
(State Key Laboratory of Non-food Biomass Enzyme Technology,National Engineering Research Center for Non-food Biorefinery,Guangxi Biomass Industrialization Engineering Institute,Guangxi Key Laboratory of Biorefinery,Guangxi Bioscience and Technology Research Center,Guangxi Academy of Sciences,Nanning,Guangxi,530007,China)
【目的】纤维素酶在生物燃料、食品、造纸、制药和纺织工业应用广泛。许多生物都能产生纤维素酶,因此纤维素酶的进化关系非常复杂;加上人类用工程手段不断改造纤维素酶,使得纤维素酶的进化关系更加复杂化。因此,本研究对纤维素酶的整体进化关系进行大规模的系统发育和统计分析。【方法】选取来自116种古菌、1 744种细菌和556种真核生物的2 416种纤维素酶进行系统进化分析。通过计算平均成对p距离反映每个分类组中的序列差异;然后将差异分为组内差异和组间差异,从数值上反映基因的垂直转移和水平转移;最后,通过进化树展示基因转移的趋势。【结果】来自不同门、纲、目、科和属的纤维素酶,其氨基酸序列的平均成对p距离差异很大。在各个系统分类中,纤维素酶的组间差异和组内差异亦存在很大不同。大规模的系统发育分析显示,大多数纤维素酶的进化经历了不同的路径和分岔,并形成相互交织在一起的群簇。【结论】本研究从统计分析和系统发育的角度提供纤维素酶的整体进化关系,不仅包括纤维素酶基因的垂直遗传和水平转移,还包括多时间和多种属进化,展示了纤维素酶进化的全貌。统计分析不仅丰富系统发育的分析结果,还为利用生物技术设计所需要的纤维素酶提供补充信息。
纤维素酶进化种属间差异种属内差异系统发育
Q349+.5,Q556+.2
A
1002-7378(2016)03-0190-19
网络优先数字出版时间:2016-05-17【DOI】10.13657/j.cnki.gxkxyxb.20160517.005
网络优先数字出版地址:http://www.cnki.net/kcms/detail/45.1075.N.20160517.1546.010.html
*国家自然科学基金项目(21466007,31400079,31460296,31560315),广西自然科学基金重点项目(2013GXNSFDA019007),广西国际科技成果引进与合作研究项目(1347004-1)和广西人才小高地建设专项基金项目资助。
**通讯作者:吴光(1956-),男,研究员,主要从事计算变异学和模型研究,E-mail:hongguanglishibahao@yahoo.com。
