脂溶性蟾毒配基的纯化工艺研究
2016-09-14王艳华段佳林卫国朱艳荣奚苗苗文爱东
王艳华 段佳林 卫国 朱艳荣 殷 英 郭 超 关 月 奚苗苗 文爱东 翁 琰
第四军医大学第一附属医院药剂科,陕西 西安 710032
药物研究
脂溶性蟾毒配基的纯化工艺研究
王艳华 段佳林 卫国 朱艳荣 殷 英 郭 超 关 月 奚苗苗 文爱东 翁 琰*
第四军医大学第一附属医院药剂科,陕西 西安 710032
目的:研究从蟾酥中提取分离高纯度蟾毒灵 (B)、华蟾酥毒基 (C)和脂蟾毒配基 (R)的纯化工艺条件。方法:以三种蟾毒配基的总含量和转移率为考察指标,采用单因素考察法,结合蟾酥药材的特点、化学成分及工艺成本等多方面的综合因素,考察蟾酥药材的提取、萃取及硅胶柱层析分离的具体工艺条件及参数。结果:蟾酥药材经10倍量(v/m)80%乙醇加热回流1h,再用10倍量 (v/m)乙酸乙酯萃取3次,最后用硅胶柱层析进行分离后,三种蟾毒配基的总含量可达92.26%,总转移率为80.54%。结论:此法能很好地从蟾酥中分离纯化蟾毒配基,大大提高了三个蟾毒配基的纯度。
蟾毒配基;含量;转移率;提取;萃取;硅胶柱层析
蟾酥是我国的传统中药材,由蟾蜍科动物中华大蟾蜍 (Bufo bufo gargarzinas Gantor)或黑眶蟾蜍(Bufo melanostictus Schneider)的皮肤腺及耳后腺分泌的白色浆液加工干燥而成[1],具有兴奋呼吸、致幻、镇痛、强心、升压、抗休克、局部麻醉、镇咳、抗肿瘤等一系列的药理活性[2],其化学成分按溶解性能不同分为脂溶性和水溶性两大类。虽然脂溶性成分仅占20%左右,但在发挥药理活性方面却起主导作用,其中又以蟾毒灵 (bufalin,B)、华蟾酥毒基 (cinobufagin,C)和脂蟾毒配基 (resibufogenin,R)含量较多、活性较强,且具有高效低毒的特点。目前蟾酥的提取工艺研究报道很多[3-6],但对蟾毒灵、华蟾酥毒基和脂蟾毒配基进行提取和纯化并提高三种蟾毒配基混合物纯度的研究并不多。研究以三种蟾毒配基的总含量和转移率为考察指标,采用单因素试验系统研究了蟾酥的提取纯化工艺,旨在得到高纯度的三种蟾毒配基混合物,为工业化生产提供了一定的参考。
1 仪器与试药
1.1 仪器 日立 D-7000高效液相色谱仪(L-7100泵、L-7420紫外检测器、L-7200自动进样器,日本Hitachi公司);HiQ sil C18色谱柱 (250 mm×4.6mm,5μm,日本KYATECH公司);AT-130柱温箱 (天 津 市 鑫 洲 科 技有 限 公 司);FA1104N电子分析天平 (上海民桥精密科学仪器有限公司);DF-101S集热式恒温磁力搅拌器(巩义市英峪予华仪器厂);FW-100高速万能粉碎机 (天津市泰斯特仪器有限公司);DZ-1BC真空干燥箱(天津市泰斯特仪器有限公司);玻璃柱(5 cm×90 cm,第四军医大学玻璃仪器制备室)。1.2 材料 蟾酥 (安徽省毫州市中药饮片厂);蟾毒灵对照品 (批号:1054-070211,中药固体制剂制造技术国家工程中心);华蟾酥毒基对照品(批号:110803-200605)、脂蟾毒配基对照品(批号110718-200507)购自中国药品生物制品检定所;柱层析用硅胶 (100-140目,200-300目)、薄层层析用硅胶G(青岛海洋化工厂)。甲醇、乙腈为色谱纯,其余试剂均为分析纯。
2 方法与结果
2.1 蟾毒配基的薄层色谱法鉴别 取蟾毒灵、华蟾酥毒基与脂蟾毒配基对照品,加甲醇分别制成每1mL含1mg的溶液,作为对照品溶液;再取蟾酥分离物细粉 12mg,加甲醇稀释并定容至10mL量瓶中,摇匀,作为供试品溶液。照薄层色谱法(《中国药典》2015版一部附录VIB)试验,吸取上述溶液各 10μL,分别点于同一硅胶 G薄层板上,以环己烷-三氯甲烷-丙酮 (4∶3∶3)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上显相同颜色的斑点。
2.2 蟾毒配基的含量测定
2.2.1 色谱条件 色谱柱:HiQ sil C18色谱柱(250 mm×4.6 mm,5μm);预柱:Fusion-RP 4 mm×3.0 mm;流动相:乙腈 -0.5%磷酸二氢钾溶液 (45∶55;pH:3.2);流速:1mL/min;检测波长:296nm;柱温:40℃;进样量:10μL;理论塔板数以蟾毒灵、华蟾酥毒基和脂蟾毒配基峰计算均不低于4000。
2.2.2 对照品溶液的制备 精密称取经五氧化二磷干燥至恒重的蟾毒灵与华蟾酥毒基对照品各3mg、脂蟾毒配基对照品5mg分别置10mL量瓶中,加甲醇超声溶解并稀释至刻度,摇匀,即得三种蟾毒配基单独的对照品储备液;分别精密量取1.0mL单独的对照品储备液,置同一个10mL量瓶中,加甲醇定容,摇匀,即得三种蟾毒配基混合的对照品储备液;精密量取 1.5mL混合对照品储备液,置5mL量瓶中,加甲醇定容,摇匀,即得理论上每 1mL分别含蟾毒灵与华蟾酥毒基9μg、脂蟾毒配基15μg的对照品溶液。
2.2.3 标准曲线的考察 分别精密量取 “2.2.1”项下三种蟾毒配基混合的对照品储备液 0.2、0.5、0.75、1.5、2.5mL置5mL量瓶中,加甲醇定容,摇匀。分别精密吸取稀释后的溶液及混合对照品储备液10μL,注入液相色谱仪,记录峰面积,以对照品溶液峰面积对浓度作图,蟾毒灵、华蟾酥毒基和脂蟾毒配基的线性关系回归方程分别为:A1=6287.7 C1-664.1,r1=0.9999;A2=6957.6 C2- 2102.2,r2=0.9997;A3= 6535.6 C3-2751.2,r3=0.9998。结果表明:蟾毒灵与华蟾酥毒基均在 1.2~30.0μg、脂蟾毒配基在 2.0~50.0μg范围内浓度与峰面积线性关系良好。
2.2.4 供试品溶液的制备 精密称定蟾酥提取物或萃取物细粉约25mg,置圆底烧失的重量,摇匀,经0.45μm滤膜过滤,分别精密量取2.0、1.5及0.5mL续滤液置10mL量瓶中,加甲醇定容,摇匀,即得。
分离物供试品溶液精密称定蟾酥分离物 5mg 置10mL量瓶中,加甲醇超声溶解并稀释至刻度,摇匀;精密量取1.0mL置10mL量瓶中,加甲醇定容,摇匀,经0.45μm微孔滤膜过滤即得。
2.3 提取工艺的考察
2.3.1 提取溶剂的选择 称取小于60目的蟾酥药材细粉 5g,分别采用 10倍量(v/m)的氯仿或70%、80%和90%乙醇加热回流1h,放冷至室温,抽滤,滤液蒸干,滤液干燥物及残渣置真空干燥箱干燥12h,按2.2项下方法测定提取物中蟾毒配基的含量。结果表明,随着乙醇浓度的增大,提取物中蟾毒配基总含量从22.01%增加到30.18%,而转移率从85.24%降到76.36%;使用索氏提取器以氯仿为溶剂进行提取时,虽然干燥物中三种蟾毒配基的总含量可达到61.89%,但提取转移率小于60%。综合考虑,乙醇为中药提取中最常用的溶剂,具有价廉、易得、安全、无毒、无污染等特点,因此本试验选择80%乙醇为提取溶剂。
2.3.2 粉末粒度的确定 在试验操作中所采用的粉碎机可在短时间内将药材粉碎到粒径小于60目,其中多数分布在80~100目,因此我们仅对60~80目,80~100目及小于100目的细粉进行了考察。称取这三种粒度的蟾酥药材细粉各 5g,采用10倍量 (v/m)80%乙醇加热回流 1h,放冷置室温,抽滤,滤液蒸干,滤液干燥物置真空干燥箱干燥12h,按“2.2”项下方法测定提取物中三种蟾毒配基的含量。结果表明:三种蟾毒配基的总含量和总提取率没有明显差异,均在 27%和 84%左右。因此确定将蟾酥药材粉碎到粒度小于60目。
2.3.3 提取回流时间的确定 称取小于60目的蟾酥药材细粉5g,采用10倍量 (v/m)80%乙醇分别加热回流0.5、1、2h,放冷至室温,抽滤,滤液蒸干,滤液干燥物置真空干燥箱干燥 12h,按2.2项下方法测定提取物中三种蟾毒配基的含量。结果表明:当提取时间小于1h时,提取物中三种蟾毒配基的纯度及转移率较低,但超过1h时,又均未有明显增加。因此确定回流时间为1h。
2.3.4 提取工艺验证 实验将蟾酥药材粉碎到粒径小于60目,称取此细粉三份,每份5g,采用10倍量 (v/m)80%乙醇加热回流1h,放至室温后,抽滤,滤液蒸干,滤液干燥物置真空干燥箱干燥12h后,按 “2.2”项下方法测定干燥物中指标性成份的含量及转移率。结果见表1。
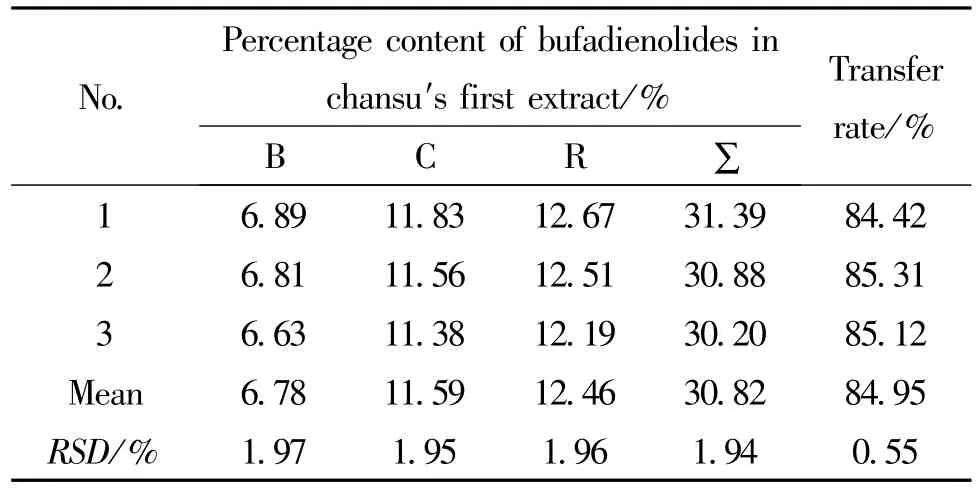
表1 蟾酥提取物的含量及转移率Tab.1 Content and transfer rateof chansu′s first extracts
2.4 萃取工艺的考察
2.4.1 萃取溶剂的选择 称取蟾酥醇提物5g,加入10倍量 (v/m)的蒸馏水超声混悬后,再分别用10倍量 (v/m)的乙醚、氯仿或乙酸乙酯萃取 3次,合并有机层并将其蒸干,所得干燥物置真空干燥箱干燥12h后,按 “2.2”项下方法测定萃取物中指标性成份的含量及转移率。结果发现:对提取物使用乙醚、氯仿或乙酸乙酯等极性较小的溶剂萃取后,蟾毒配基的总含量提高了近一倍,均在55%左右,但乙醚和氯仿的萃取转移率均小于80%,而乙酸乙酯可达到90%以上。加之使用乙醚极易发生乳化现象,同时考虑到溶剂的毒性,最终选择乙酸乙酯作为萃取溶剂,进一步考察其用量。
2.4.2 萃取溶剂用量的考察 称取蟾酥醇提物 5g,加入10倍量 (v/m)的蒸馏水超声混悬后,再分别用5、10、20倍量 (v/m)的乙酸乙酯萃取3次,合并有机层并将其蒸干,所得干燥物置真空干燥箱干燥12h后,按 “2.2”项下方法测定萃取物中指标性成份的含量及转移率。结果表明:使用不同用量的乙酸乙酯均能有效地对指标性成分进行萃取,但在操作中使用五倍量的蒸馏水混悬醇提物较困难,固状物不能充分混悬,操作不便,同时考虑到节省溶剂,最终选用10倍量的乙酸乙酯进行萃取。
2.4.3萃取工艺验证 实验称取蟾酥醇提物 20g,加入10倍量 (v/m)的蒸馏水超声混悬后,再分别用10倍量 (v/m)的乙酸乙酯萃取 3次,合并有机层并将其蒸干,所得干燥物置真空干燥箱干燥12h后,按“2.2”项下方法测定萃取物中指标性成份的含量及转移率,结果见表2。

表2 蟾酥萃取物的含量及转移率Tab.2 Content and transfer rateof chansu′s second extracts
2.5 硅胶柱层析工艺的考察
2.5.1 柱层析条件 自制玻璃 (D-5cm,H-8 cm),玻璃柱 (5cm×90cm),层析用硅胶,展开剂:环己烷、丙酮、三氯甲烷。
2.5.2 硅胶柱装柱及层析分离 称取 250g硅胶(200~300目),用环己烷分散均匀后,装柱并沉降过夜。称取蟾酥萃取物10g溶于150mL氯仿与甲醇(1∶1)中,滤去不溶物,将澄清液边加边搅拌于50g硅胶 (100~140目)中拌样,放置待完全干燥后,装入已经沉降一晚的硅胶柱顶,上面再加入保护硅胶和无水硫酸钠。洗脱时,先用环己烷:丙酮(10∶1)洗脱3个体积,再用环己烷:丙酮 (5∶1)洗脱5个体积,然后用丙酮洗脱3个体积,最后用甲醇洗脱至目标成分消失。洗脱过程中,分段收集馏分,利用减压蒸馏对馏分进行浓缩,并用薄层色谱法对浓缩物进行鉴别。由鉴别结果可知伴随第二个黄色带洗脱下来时,开始得到的白色固体即为指标成分。分离结束后合并经分析较纯的蟾毒灵、华蟾酥毒基和脂蟾毒配基,并对合并物按 “2.2”项下方法进行定量分析,结果见表3。

表3 蟾酥分离物的含量及转移率Tab.3 Content and transfer rateof chansu′s separated products
3 讨论
3.1 提取方法的确定 国内虽有多篇文献报道了蟾酥中蟾毒配基的提取方法,但多数是为了分离少量试验样品,因此考虑不是很全面,存在着各种问题。研究综合文献的优缺点,使用回流提取法以乙醇为溶剂对蟾酥中脂溶性成分进行提取。
该法简便,省时,溶剂用量较少,同时使用萃取工艺对蟾毒配基进行富集,最终萃取物中三种目标化合物的纯度和在50%以上,转移率在90%以上,为工业化放大奠定了一定实验基础。
3.2 纯化工艺的选择 蟾毒灵、华蟾酥毒基及脂蟾毒配基属于强心苷成分,极性较小不适合用大孔树脂来分离。若将这三种蟾毒配基的纯品于环己烷 -丙酮中用中性氧化铝吸着,经1~3d即起吸附剂副反应,综合比较使用硅胶柱分离效果较好[7]。研究使用湿柱层析法,进行梯度洗脱,可一次性将三种蟾毒配基从萃取物中进行分离,最终蟾酥分离物中三种蟾毒配基的含量和达到了92.26%,转移率为 80.54%。在实验室工艺放大过程中,可适量增加上样量,减少分离硅胶的使用,多次层析分离后可将每根层析柱的少量交叉部分合并,再次上柱层析分离,可使纯化效率提高同时也保证了蟾毒配基的转移率。
[1]Y.Chen,J.Xiang,W.Gu,et al.Chemical constituents of Bufo siccus [J].中国中药杂志,1998,23(10):620-640.
[2]K.K.Chen,A.Kovax:Pharmacology and toxicology of toad venom[J]. J.Pharm.Sci.1967,56,1535-1541.
[3]李妍,王四旺,孙纪元,等.正交试验法优选蟾酥脂溶性成分提取工艺[J].西北药学杂志,2006,22(3):112-113.
[4]赵大洲,邱鹰昆,陈继永,等.大孔树脂富集蟾皮总二烯内酯的工艺研究 [J].时珍国医国药,17(10):1991-1992.
[5]乔莉,段文娟,姚遥,等.蟾酥中强心甾类化学成分的分离与鉴定[J].沈阳药科大学学报,2007,24(10):593-596.
[6]霍建中,孙云霞.蟾酥的分离及微量组分分析 [J].天津化工,1997,10(1):39-40.
[7]徐任生,陈仲良.中草药有效成分提取与分离 [M].上海:上海科学技术出版社,1983.
Study on the purification technology of high purity of liposoluble bufadienolides
WANG Yanhua DUAN Jialin WEIGuo ZHU Yanrong YIN Ying GUO Chao
GUAN Yue XI Miaomiao WEN Aidong WENG Yan*
Department of Pharmacy,Xijing Hospital,Fourth Military Medical University,Xi'an 710032,China
Objective To study on the purification technology of Chansu in order to obtain the high purity ofbufalin(B),cinobufagin(C)and resibufogenin(R).Methods The single factor investigation method,combining with comprehensive factors such as the characteristics of raw material,chemical composition and technological cost was used to investigate the detailed conditions and parameters of extraction,separation and silica gel column chromatographyltechnologies with the content and the transfer rate of three bufadienolides as index.Results The final separated products contained a high purity of 92.26% for a sum of B,C and R and a high total transfer rate of80.54%after the raw materials were separated by 10-fold(v/m)80%ethanol reflux extraction for 1h,10-fold(v/ m)ethyl acetate extraction for3 times and loaded on silica gel columnchromatography.Conclusion Bufadienolides could be well separated from the raw material of Chansu,and the purityof bufadienolides could be greatly improved with this method.
bufadienolides;content;transfer rate;extraction;separation;silica gel column chromatography
R284.2
A
1007-8517(2016)13-0015-03
2016.05.03)
国家自然科学基金 (81303264)。
王艳华 (1987-),女,硕士,药师,主要从事中药新制剂的研究与开发。E-mail:645301987@qq.com
翁琰,药师,博士。E-mail:mejean12021984@aliyun.com
