三元金属硫化物-石墨相氮化碳异质结催化剂的制备及光催化性能
2016-09-13王彦娟孙佳瑶封瑞江张健
王彦娟 孙佳瑶 封瑞江 张健
(辽宁石油化工大学化学化工与环境学部,辽宁抚顺113001)
三元金属硫化物-石墨相氮化碳异质结催化剂的制备及光催化性能
王彦娟孙佳瑶封瑞江张健*
(辽宁石油化工大学化学化工与环境学部,辽宁抚顺113001)
以双氰胺、醋酸锌、钼酸铵、醋酸镉和硫化钠为原料,采用水热法合成了一系列Zn-Mo共掺杂CdS (Zn-Mo-CdS),并与g-C3N4组成异质结催化剂(Zn-Mo-CdS/g-C3N4)。采用X射线衍射光谱(XRD)、紫外-可见(UV-Vis)光谱、电感耦合等离子体-原子发射光谱(ICP-AES)、电化学阻抗谱(EIS)、X光电子能谱(XPS)等分析手段对制备的催化剂进行了表征。结果表明,Zn-Mo-CdS与g-C3N4之间紧密结合并形成异质结,促进界面电荷迁移,抑制光生电子-空穴对的复合。以可见光下降解染料罗丹明B(RhB)为探针反应考察了催化剂性能。结果表明,Zn-Mo-CdS/g-C3N4异质结催化剂的光催化性能与单纯g-C3N4、Zn-Mo-CdS及双金属硫化物/ g-C3N4异质结催化剂相比均有大幅度提高,质量比m(Zn-Mo-CdS)/m(g-C3N4)=4:1时制备的异质结催化剂表现出最大的降解速率常数,是单纯g-C3N4和Zn-Mo-CdS的30倍和10倍。不仅Zn-Mo-CdS,其他三元金属复合硫化物如Mo-Ni-CdS和Ni-Sn-CdS与g-C3N4之间也能有效构筑异质结,促进电子-空穴对的分离和催化性能提升。
g-C3N4;三元金属硫化物;异质结;光催化;有机物降解
1 引言
近年来,日益严重的环境污染和能源危机使得人们对清洁能源的渴望越发强烈。太阳能作为清洁能源的代表被认为是最佳的传统化石能的替代者。以太阳能为能源的光催化技术能驱动一系列化学反应,将太阳能转化为化学能,这一技术已经在解决环境污染和能源危机方面表现出巨大的潜力。目前,最常用的光催化剂为金属氧化物,如TiO2、ZnO等。这些传统光催化剂带隙能较大,对可见光的吸收利用率很差,大大降低了太阳能在现实中的应用前景1,2。
最近,有机非金属材料石墨相氮化碳(g-C3N4)引起了来自物理、化学、材料、环保等领域众多学者的关注。g-C3N4的带隙能较窄(2.7 eV),可直接吸收波长小于460nm的可见光。此外,g-C3N4还具有良好的化学稳定性、热稳定性以及独特的电子结构,因此在光催化分解水制氢3、有机合成4、分解有机污染物5等领域有广泛的应用前景。但是,由于g-C3N4的光生电子和空穴易复合,大大抑制了其光催化性能6。
开发异质结光催化剂是解决上述问题的有效途径之一。光生电荷能通过异质结快速地转移,有效抑制复合。Zhang等7采用自组装和化学沉淀法制备了g-C3N4/Bi2O2CO3层状异质结催化剂,发现制备方法对两组分间的结合强度有很大影响,进而影响电子-空穴分离效率及光催化性能。Ong等8制备了氧化石墨烯(GO)/g-C3N4异质结催化剂并用于还原CO2制甲醇。研究发现GO与g-C3N4之间以C―O―C键形式链接,大大提高了催化剂的量子效率。Ong等9还制备了质子化g-C3N4,并通过正负电荷间的静电作用制备了还原氧化石墨烯(rGO)/质子化g-C3N4复合催化剂,进一步提高了催化剂量子效率。在可见光下还原CO2制甲醇的反应中,rGO/质子化g-C3N4复合催化剂的甲醇产率分别是纯g-C3N4和rGO/未质子化g-C3N4异质结催化剂的5.4倍和1.7倍。除此之外,多种石墨相氮化碳基异质结催化剂如g-C3N4/BiIO410、g-C3N4/ MoO311、g-C3N4/Bi2WO612等也相继被报道。
合理设计异质结对提高光催化性能有显著影响,而设计异质结最重要的依据是组分间的能级匹配关系。近年来,纳米结构的金属硫化物在光催化领域备受关注,主要有单金属组分硫化物ZnS、CdS、In2S3、SnS213-16等,双金属组份硫化物ZnIn2S416、ZnxCd1-xS17、CuInS218等。这些金属硫化物一般具有较窄的禁带宽度,能够吸收可见光。另一方面,多种金属硫化物的价带与导带的能级位置与g-C3N4的能级位置相互匹配19,能满足形成异质结催化剂的基本条件。因此,许多研究者致力于制备金属硫化物/g-C3N4异质结催化剂来提高光生电子-空穴对的分离速率。Ge等20采用浸渍-沉积法合成了g-C3N4/MoS2异质结催化剂,XPS分析表明层状MoS2很好地镶嵌在g-C3N4表面并形成异质界面,有效促进了光生电荷转移。当MoS2占催化剂总质量0.5%时,催化剂在可见光下表现出最佳的分解水制氢气性能,析氢速率为23.10μmol∙h-1,是纯g-C3N4的11.3倍。Sun等21合成了g-C3N4/ ZnCdS异质结催化剂,并用于还原水中的Cr(VI)离子和降解染料RhB。结果表明,m(g-C3N4)/ m(ZnCdS)=1:4(CNZS-20)时,催化剂表现出最佳的催化性能,45m in对RhB的降解率高达99%。通过高倍透射电镜观察到制备的异质结催化剂中具有紧密结合的两种不同晶面,说明在g-C3N4/ ZnCdS催化剂内部形成了异质结界面,为电荷转移提供良好的条件。同时,CNZS-20复合催化剂在25m in内对水中的Cr(VI)离子还原率达到99%。Sun等22采用溶剂热法制备了三维(3D)结构花状g-C3N4/SnS2催化剂,并用于可见光下还原水中的Cr (VI)离子。结果表明,异质结催化剂优越的催化性能是由于改善了催化剂的电子-空穴分离效率以及两组分间匹配的能级结构。上述方法虽然从一定程度上提高了光生载流子寿命及电子空穴对的分离效率,然而科学技术水平日新月异的发展迫使科研工作者们对催化剂的性能提出了更高的要求。
据我们所知,目前的中外研究报道中还未见有关三金属组份硫化物/g-C3N4异质结催化剂的性质与光催化性能方面的研究报道。MoS2是层状金属硫化物,作为助催化剂可以提高电子-空穴分离效率,被认为是贵金属Pt的最佳替代者23-25。ZnS则是最常用的光催化剂之一,其电子结构和光学性质可调,氧化降解有机物能力强26,27。本文以双氰胺、醋酸锌、钼酸铵和醋酸镉和硫化钠为原料,采用水热法制备了一系列Zn-Mo-CdS/g-C3N4异质结催化剂,考察了催化剂在可见光下对染料RhB的光降解性能。系统地探讨了异质结组成对催化剂结构性质、光学性质以及光催化性能的影响。结果表明,m(Zn-Mo-CdS)/m(g-C3N4)=1:4时,异质结催化剂表现出最佳的光催化性能,速率常数达到0.06m in-1,是单纯g-C3N4和Zn-Mo-CdS的30倍和10倍。相似的三金属组份硫化物/g-C3N4异质结催化剂如Ni-Sn-CdS/g-C3N4和Mo-Ni-CdS/g-C3N4也表现出优越的光催化性能。
2 实验部分
2.1Zn-Mo-CdS/g-C3N4异质结催化剂的制备
所用试剂均为分析纯,购自天津市富宇精细化工有限公司。根据文献方法,采用双氰胺为前驱体制备g-C3N428。称取一定量醋酸锌、钼酸铵与醋酸镉(n(Zn)/n(Mo)/n(Cd)=1:1:8)溶于10m L去离子水中得到溶液,搅拌15m in后,一定量的g-C3N4(0.1,0.2,0.5,1.6g)加入上述溶液,超声分散30m in,搅拌条件下缓慢滴加20m L浓度为0.5mol∙L-1的硫化钠溶液。搅拌12 h后,装反应釜160°C合成16h。待反应釜冷却至室温后将产物离心分离,用去离子水和无水乙醇洗涤数次,80°C下过夜干燥。得到一系列Zn-Mo-CdS/g-C3N4异质结复合催化剂,记为ZMCS-CN(w%),其中w%为g-C3N4的质量分数。重复上述制备过程。在不加g-C3N4条件下,得到的催化剂命名为Zn-Mo-CdS。为了分析Zn-Mo共掺杂的作用,采用相同的制备方法制备纯CdS作对比。上述合成方法及金属摩尔比不变,将三种金属盐两两组合,制备的催化剂命名为ZnMoS-CN(20%),ZnCdS-CN(20%)和MoCdS-CN(20%)。
2.2结构表征
X射线衍射光谱(XRD)采用日本岛津公司的X射线衍射仪(XRD-7000)进行测定,以Cu靶Kα1作为辐射电源,工作电压40kV,工作电流30mA。采用扫描电子显微镜(SEM,JSM5600LV,日本电子有限公司)对制备的催化剂形貌进行表征。紫外-可见(UV-Vis)光谱采用日本JASCA公司的紫外-可见光谱仪(UV-550)进行测定。X光电子能谱(XPS)使用赛默飞世尔科技有限公司的Thermo ESCALAB 250光电子能谱仪进行测定。电化学阻抗谱采用AUTO-LAB PGSTAT30电化学工作站(瑞士万通公司)进行测定。测试在自制带石英窗口的电解池中进行(20nm×40nm×50mm),体系采用三电极体系,以膜状样品为工作电极,Pt片为对电极,Ag/AgCl为参比电极,扫描频率范围为1-100MHz,控制在开路电位,扰动电位为l0m V,电解质为0.5mol∙L-1Na2SO4。采用上海辰华CHI760C型电化学工作站测试样品的光电流,500W氙灯为光源,0.5mol∙L-1Na2SO4为电解质。
2.3活性评价
以250W高压钠灯(主波长400-800nm)为可见光光源,以浓度为0.5mol∙L-1的亚硝酸钠水溶液为钠灯冷却循环水滤去光源中紫外光部分。以染料RhB为降解物评价催化剂的光催化活性。0.1 g催化剂加入到500m L浓度为10mg∙L-1的RhB中,搅拌0.5h达到催化剂对反应底物的吸附平衡。搅拌下将溶液放在光源下进行照射,同时鼓入空气(30°C,标准大气压)。每隔0.5h取一个样品置于离心管中,在转速为3000r∙m in-1的条件下离心5m in后取上层清液,用紫外-可见分光光度计于550nm测定吸光度。
3 结果和讨论
3.1XRD表征
图1为g-C3N4、Zn-Mo-CdS和ZMCS-CN(w%)的XRD图谱。g-C3N4有两个特征峰,分别位于2θ为13.1°和27.5°处,其中27.5°处的衍射峰最强,为芳香物层间堆积特征峰,晶面指数标记为(002),对应层间距d=0.33 nm,说明g-C3N4具有和石墨类似的层状结构。13.1°处的衍射峰是melon类物质的特征峰,晶面指数标记为(100),对应3-s-三嗪结构中氮孔结构,间距d=0.67 nm29。CdS在2θ位于26.6°、43.9°和52°处出现的3个较强的衍射峰,为立方相CdS的特征峰,分别对应(111)、(220)、(311)晶面30。Zn-Mo-CdS有三个特征峰,分别位于2θ为26.8°、44.0°和52.2°处,与CdS的特征峰位置非常相近。此外,谱图中没有发现MoS2和ZnS的特征衍射峰22,31。这可能是由于Zn和Mo掺入了CdS的晶格得到了三元金属复合硫化物Zn0.1Mo0.1Cd0.8S1.1,而非单纯的ZnS,MoS2和CdS的混合物。正是由于Zn和Mo的掺杂作用使得CdS发生晶格扭曲,使衍射峰位置略有偏移。与Zn-Mo-CdS相比,制备的g-C3N4/Zn-Mo-CdS异质结催化剂的衍射峰位置没有发生变化。g-C3N4/Zn-Mo-CdS异质结催化剂中没有明显的g-C3N4的衍射锋。这可能是由于CdS在26.6°处的衍射峰与g-C3N4在27.5°处的衍射峰相互叠加导致g-C3N4的衍射锋被屏蔽。Ge等32制备了g-C3N4/Bi2WO6异质结催化剂,Bi2WO6的XRD特征峰(28.2°)与g-C3N4的特征峰位置十分接近,在g-C3N4的含量低于50%(w)时也表现出类似的屏蔽现象。
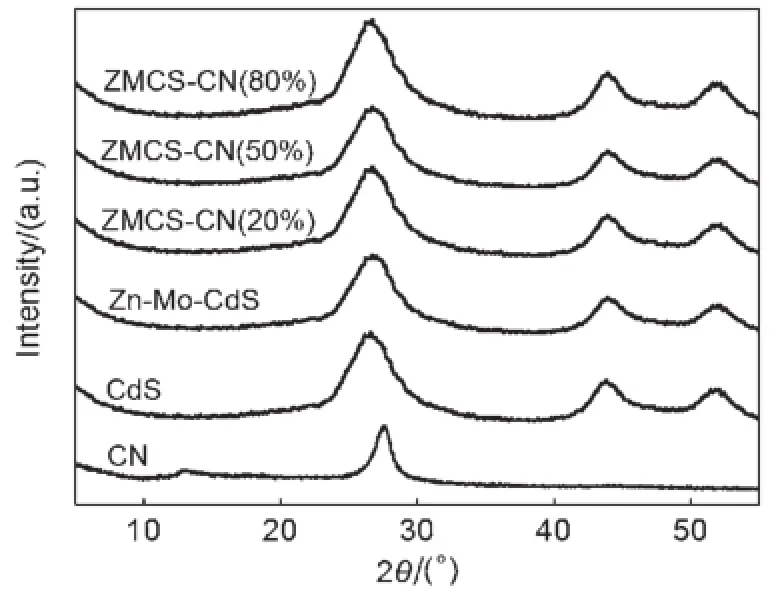
图1 CN,CdS,Zn-Mo-CdS和ZMCS-CN(w%)的XRD图谱Fig.1 XRD patternsof CN,CdS,Zn-Mo-CdS, and ZMCS-CN(w%)
3.2SEM结果
采用扫描电子显微镜(SEM)对制备催化剂形貌进行分析(图2)。如图2a所示,制备的g-C3N4具有无规则的类似于石墨的光滑层状结构。图2b所示的是制备的Zn-Mo-CdS,从图中可以清晰地看出, Zn-Mo-CdS为大小均一的椭圆形纳米颗粒,其直径约为50nm。图2c为制备的ZMCS-CN(20%)催化剂。与纯g-C3N4相比,ZMCS-CN(20%)表面非常的粗糙。由此判断,Zn-Mo-CdS纳米颗粒均匀地附着在了g-C3N4的表面。这种面-面接触方式有利于形成异质结,可促进界面电荷迁移,抑制光生电子-空穴对的复合。ICP结果显示,Zn、Mo、Cd和S的质量分数分别为4.0%、5.6%、50.2%和20.2%,与理论值非常接近。
3.3UV-Vis结果
制备催化剂的UV-Vis光谱如图3所示。g-C3N4的吸收边界约为460nm,利用公式Eg=1240/λg(Eg为禁带宽度,λg为吸收波长阈值)计算出其带隙能约为2.69 eV,与文献报道结果相一致。CdS的吸收边界为556nm,对应能带为2.23 eV。Zn-Cd共掺杂后,Zn-Mo-CdS的吸收边界略微蓝移,对应的带隙能为2.25eV。制备的g-C3N4/Zn-Mo-CdS复合催化剂的吸收边界介于g-C3N4和Zn-Mo-CdS之间,证明了两组分间存在电子耦合作用并由此形成异质结。Liu33和Dong34等也报道了类似的实验结果。除此之外,Zn-Mo-CdS/g-C3N4异质结催化剂在全波长范围内的吸收强度都要高于g-C3N4。这说明制备的Zn-Mo-CdS/g-C3N4异质结催化剂比单纯g-C3N4催化剂吸收更多的光能,产生更多的光生电子空穴对,因此更有利于光催化反应发生。

图2 CN(a),Zn-Mo-CdS(b)和ZMCS-CN(20%)(c)的SEM图片Fig.2 SEMim agesof CN(a),Zn-Mo-CdS(b),and ZMCS-CN(20%)(c)
3.4电子-空穴迁移示意图
根据文献报道,半导体材料的价带能级(EVB)可以利用公式EVB=X-E-1/2Eg计算得出,其中X为半导体的绝对电负性,E为氢标下自由电子对的能量(约为4.5eV),Eg为半导体带隙能35。计算结果显示,Zn-Mo-CdS的X值为5.21 eV,EVB为1.84eV。相应的导带位置ECB位于-0.41 eV。g-C3N4的ECB和EVB分别位于-1.12 eV和+1.57 eV36。由此可见,Zn-Mo-CdS和g-C3N4的能级匹配完好。当二者组成异质结催化剂后,在电势差驱动力的作用下,光电子能快速地从g-C3N4转移至Zn-Mo-CdS表面,而空穴会从Zn-Mo-CdS向g-C3N4聚集。电子和空穴重新分布后,会在两组分间的异质界面建立一个稳定的内电场,抑制电子-空穴的复合过程,使量子效率和光催化性能得到极大提升,如图4所示。
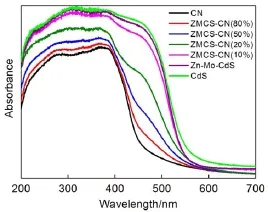
图3 CN、CdS、Zn-Mo-CdS和ZMCS-CN(w%)的UV-Vis图谱Fig.3 UV-Vis spectra of CN,CdS,Zn-Mo-CdS, and ZMCS-CN(w%) coloronweb version

图4 制备异质结催化剂Zn-Mo-CdS/g-C3N4的电子-空穴迁移示意图Fig.4Schematic diagram of electrons-holesm igration over as-prepared Zn-Mo-CdS/g-C3N4heterojunction photocatalyst
3.5XPS表征
X射线光电子能谱是表征催化剂表面元素状态的有效手段。通常来讲,电子密度越低,原子核对核外电子的束缚能力越强,相应的结合能就越高。图5为制备催化剂的C 1s,N 1s,S 2p,Zn 2p,Mo 3d和Cd 3d能级的XPS图谱。如图5(a)所示,CN的C 1s图谱可分为三个峰,其中结合能为284.6eV的峰归属为环状结构sp2杂化的C原子(N―C=N),286eV的峰归属为sp3杂化的C原子(C―(N)3),288.4eV的峰归属为石墨型结构中的C―N键中的C原子。图5(b)中CN的N 1s图谱可分为两个峰,其中结合能为398.4eV的峰归属为sp2杂化的N原子(C―N=C),400.7 eV的峰归属为芳香环中连接三个C原子的N原子37。ZMCS-CN (20%)与CN相比,C 1s轨道的三个峰以及N 1s轨道在398.4eV的峰均向高结合能方向偏移,这可能是由于富电子的g-C3N4将部分电子转移至Zn-Mo-CdS表面,导致自身电子密度降低引起的。此结果证实了g-C3N4与Zn-Mo-CdS之间存在较强的相互作用。如图5c所示,Zn-Mo-CdS样品中的S 2p轨道的结合能位置位于162.1 eV处,说明存在M(金属)―S键,硫的价态为S2-。ZMCS-CN(20%)样品中,除了M―S键,硫还表现出另外一种存在形式,其结合能位于163.3 eV处。此处结合能在Zn-Mo-CdS样品中并没有显示,说明此结合能与氮化碳的复合有直接关系,复合后Zn-Mo-CdS中的部分S元素受到氮化碳的影响化学环境发生改变导致产生此结合能,由此也证实了Zn-Mo-CdS与氮化碳是紧密结合而非单纯混合的。
图5(c,d,e)所示为Zn 2p,Mo 2p和Cd 3d轨道的XPS谱图。Zn-Mo-CdS中的Zn 2p轨道的结合能位置位于1021.7和1044.9 eV处,Mo 2p轨道的结合能位于230.2和233.4eV处,Cd 3d轨道的结合能位于404.7和411.5eV处,与单纯的ZnS、MoS2和CdS在相应轨道处的结合能有显著区别20,38-41。这也再次证实了上述的结论:制备的催化剂不是单纯的ZnS、MoS2和CdS的混合物,而是三元金属复合硫化物Zn0.1Mo0.1Cd0.8S1.1。金属间的相互作用导致了结合能的差异。对于ZMCS-CN(20%)、Zn 2p、Mo 2p和Cd 3d轨道的结合能与Zn-Mo-CdS相比均向低结合能方向发生明显的蓝移现象。这一结果再次证实了g-C3N4与Zn-Mo-CdS之间存在较强的相互作用。
3.6EIS表征
图6为制备的催化剂的电化学阻抗谱图。在Nyquist曲线中,阻抗弧半径越小表明电子转移速率越快,光生电子空穴对的复合速率越低42,43。从图中可知,CN的圆弧半径最大,表明其电荷转移电阻大,不利于电荷传输。Zn-Mo-CdS的阻抗弧半径小于CdS,说明Zn-Mo共掺杂提高了电子-空穴的分离效率,这可能是由于掺杂引起了CdS的晶面缺陷导致的。异质结催化剂的阻抗弧半径进一步减小。其中,ZMCS-CN(20%)的阻抗弧半径最小,表示该催化剂具有最高效的界面电荷迁移速率,能最有效的分离光生电子-空穴对。

图5 制备催化剂的C 1s(a),N 1s(b),S 2p(c),Zn 2p(d),Mo 3d(e)和Cd 3d(f)能级的XPS图谱Fig.5XPS spectra of as-p repared catalysts in the region of C 1s(a),N 1s(b),S 2p(c),Zn 2p(d),Mo 3d(e),and Cd 3d(f)
3.7光电流
图7为制备催化剂的光电流响应结果。可以看出Zn-Mo-CdS的光电流强度大于CN,而ZMCSCN(20%)的光电流明显强于两个组分,约为Zn-Mo-CdS的2.5倍,且衰减现象不明显。结合EIS表征结果可以证实Zn-Mo-CdS和g-C3N4间紧密结合形成异质结,提高了电子-空穴对的分离效率,而非简单的物理混合。
3.8催化性能评价
CN、Cd、Zn-Mo-CdS和ZMCS-CN(w%)在可见光下对RhB的降解反应结果如图8a所示。暗态吸附结果表明0.5h后所有催化剂对反应底物可以达到吸附平衡。CN在60m in内对RhB的降解率为17%。CdS与Zn-Mo-CdS的活性分别为26%和40.5%,说明掺杂后CdS的光催化性能大幅提升。ZMCS-CN(w%)系列催化剂的光催化性能与g-C3N4和Zn-Mo-CdS相比均有显著提高,这是由于g-C3N4与Zn-Mo-CdS复合后形成异质结界面,促进了光生电子-空穴对的有效分离,大大提高了量子效率。ZMCS-CN(20%)展示了最佳的光催化性能,在60m in内对RhB的降解率达到98%。此外,ZMCS-CN(w%)系列催化剂对RhB的吸附能力与两个单组分催化剂相比有显著提高。反应底物的吸附是多相催化的重要环节,吸附能力的提升对催化剂的性能提升也有着显著的促进作用。
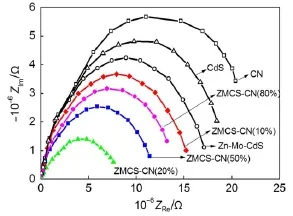
图6 制备催化剂的电化学阻抗谱Fig.6EISspectra of as-prepared catalysts
通常情况下,光降解反应遵循一级反应动力学,动力学方程为:-ln(C/C0)=kt,其中C和C0分别代表任意时刻和初始时刻RhB的浓度,由直线的斜率可得到速率常数k44。如图8(a)的插图所示,经过计算,CN和Zn-Mo-CdS的速率常数分别为0.002和0.006m in-1。复合催化剂ZMCS-CN(10%),ZMCS-CN(20%),ZMCS-CN(50%)和ZMCS-CN (80%)的速率常数分别为0.03、0.06、0.04和0.034m in-1。ZMCS-CN(20%)表现出最大的速率常数,是单纯g-C3N4和Zn-Mo-CdS的30倍和10倍。图8 (b)对比了ZMCS-CN(20%)、ZnMoS-CN(20%)、ZnCdS-CN(20%)和MoCdS-CN(20%)降解RhB的光催化性能。可以看出ZnMoS-CN(20%),ZnCdSCN(20%)和MoCdS-CN(20%)的活性差别不大,60m in的降解率均在40%-50%之间,明显低于ZMCS-CN(20%)。由此可见,三组分金属硫化物与g-C3N4组成的异质结催化剂比之双组分金属硫化物与g-C3N4组成的异质结催化剂催化性能有显著提升。

图7 g-C3N4基催化剂的光电流响应Fig.7 Photocurrent responsesof g-C3N4based catalysts

图8 制备的催化剂在可见光下降解RhB的催化性能评价及其降解一级反应动力学Fig.8 Photocatalytic performancesof RhB degradation for as-prepared catalystsunder visib le light ir rad iationsand their first-order k inetics
此外,采用相似的水热法和金属摩尔比,制备了其他两种三元金属复合硫化物Mo-Ni-CdS和Ni-Sn-CdS及相应的异质结催化剂Mo-Ni-CdS-CN (20%)和Ni-Sn-CdS-CN(20%)。可见光下对RhB的降解反应结果表明(图9),Mo-Ni-CdS-CN(20%)和Ni-Sn-CdS-CN(20%)的催化性能均优于相应的单组分催化剂,速率常数分别为0.026和0.015m in-1,是相应三元金属硫化物的5.3倍和4.2倍,是纯g-C3N4的13倍和7.5倍。此结果说明不仅Zn-Mo-CdS,其他三元金属硫化物与g-C3N4之间也能有效构筑异质结,促进电子-空穴对的分离。
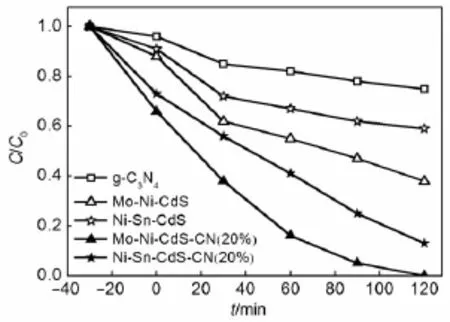
图9 MoNiCdS-CN(20%)和NiSnCdS-CN(20%)在可见光下的光催化性能Fig.9 Photocatalytic per formancesofMo-Ni-CdS-CN (20%)and Ni-Sn-CdS-CN(20%)under visib le light
4 结论
以双氰胺、醋酸锌、钼酸铵、醋酸镉和硫化钠为原料,采用水热法成功制备了一系列Zn-Mo-CdS/g-C3N4异质结催化剂。Zn-Mo-CdS与g-C3N4之间紧密结合,构筑异质结,促进界面电荷迁移,抑制光生电子-空穴对的复合,使量子效率和光催化性能得到极大提升。在可见光下对RhB的降解反应结果显示,Zn-Mo-CdS/g-C3N4异质结催化剂的光催化性能与单纯g-C3N4和Zn-Mo-CdS相比均有显著提高,ZMCS-CN(20%)表现出最大的速率常数(0.06m in-1),是单纯g-C3N4和Zn-Mo-CdS的30倍和10倍。不仅Zn-Mo-CdS,其他三元金属复合硫化物如Mo-Ni-CdS和Ni-Sn-CdS与g-C3N4之间也能有效构筑异质结,促进电子-空穴对的分离和催化性能提升。
References
(1)Kondo,K.;Murakami,N.;Ye,C.;Tsubota,T.;Ohno,T.Appl. Catal.B:Environ.2013,142-143,362.
(2)Chen,X.B.;Shen,S.H.;Guo,L.J.Chem.Rev.2010,110, 6503.doi:10.1021/cr1001645
(3)Zhang,G.G.;Zhang,M.W.;Ye,X.X.;Qiu,X.Q.;Lin,S.; Wang,X.C.Adv.Mater.2014,26,805.doi:10.1002/ adma.201303611
(4)Xu,J.;Wu,H.T.;Wang,X.;Xue,B.;Li,Y.X.;Cao,Y.Phys. Chem.Chem.Phys.2013,15,4510.doi:10.1039/c3cp44402c
(5)Ge,L.Mater.Lett.2011,65,2652.doi:10.1016/j. matlet.2011.05.069
(6)Niu,P.;Zhang,L.;Liu,G.;Cheng,H.Adv.Funct.Mater.2012, 22,4763.doi:10.1002/adfm.v22.22
(7)Zhang,Q.;Wang,H.Y.;Hu,S.Z.;Lu,G.;Bai,J.;Kang,X. X.;Liu,D.;Gui,J.Z.RSC Adv.2015,5,42736.doi:10.1039/ C5RA 04189A
(8)Ong,W.J.;Tan,L.L.;Chai,S.P.;Yong,S.T.Chem.Commun. 2015,51,858.doi:10.1039/C4CC08996K
(9)Ong,W.J.;Tan,L.L.;Chai,S.P.;Yong,S.T.;Mohamed,A. R.Nano Energy 2015,13,757.doi:10.1016/j. nanoen.2015.03.014
(10)Tian,N.;Huang,H.W.;He,Y.;Guo,Y.X.;Zhang,Y.H.RSC Adv.2014,4,42716.doi:10.1039/C4RA05917D
(11)He,Y.M.;Zhang,L.H.;Wang,X.X.;Wu,Y.;Lin,H.J.;Zhao, L.H.;Weng,W.Z.;Wan,H.L.;Fan,M.H.RSC Adv.2014,4, 13610.doi:10.1039/c4ra00693c
(12)Wang,Y.J.;Bai,X.J.;Pan,C.S.;He,J.;Zhu,Y.F.J.Mater. Chem.2012,22,11568.doi:10.1039/c2jm16873a
(13)Hu,J.S.;Ren,L.L.;Guo,Y.G.;Liang,H.P.;Cao,A.M.; Wan,L.J.;Bai,C.L.Angew.Chem.Int.Edit.2012,44,1269.
(14)Yan,H.J.;Yang,J.H.;Ma,G.J.;Wu,G.P.;Zong,X.;Lei,Z. B.;Shi,J.Y.;Li,C.J.Catal.2009,266,165.doi:10.1016/j. jcat.2009.06.024
(15)Huo,Y.N.;Yang,X.L.;Zhu,J.;Li,H.X.Appl.Catal.B: Environ.2011,106,69.
(16)Fan,Y.H.;Luo,Q.;Liu,G.X.;Wang,J.X.;Dong,X.T.;Yu, W.S.;Sun,D.Chin.J.Inorg.Chem.2014,30,627.[范英华,雒琴,刘桂霞,王进贤,董相廷,于文生,孙德.无机化学学报,2014,30,627.]
(17)Nie,Q.L.;Yuan,Q.L.;Wang,Q.S.;Xu,Z.D.J.Mater.Sci. 2004,39,5611.doi:10.1023/B:JMSC.0000039301.70811.a4
(18)Xia,S.;Lei,W.;Yang,Y.L.Nanoscale Res.Lett.2011,6, 562.doi:10.1186/1556-276X-6-562
(19)Xu,Y.;Schoonen,M.A.A.Am.Mineral.2000,85,543.doi: 10.2138/am-2000-0416
(20)Ge,L.;Han,C.;Xiao,X.Int.J.Hydrog.Energy 2013,38, 6960.doi:10.1016/j.ijhydene.2013.04.006
(21)Sun,M.;Yan,T.;Yan,Q;Liu,H.Y.;Yan,L.G.;Zhang,Y.F.; Du,B.RSCAdv.2014,4,19980.doi:10.1039/c4ra01439a
(22)Sun,M.;Yan,Q.;Yan,T.;Li,M.M.;Wei,D.;Wang,Z.P.; Wei,Q.;Du,B.RSC Adv.2014,4,31019.doi:10.1039/ C4RA03843F
(23)Xiang,Q.;Yu,J.;Jaroniec,M.J.Am.Chem.Soc.2012,134, 6575.doi:10.1021/ja302846n
(24)Zong,X.;Yan,H.J.;Wu,G.P.;Ma,G.J.;Wen,F.Y.;Wang, L.;Li,C.J.Am.Chem.Soc.2008,130,7176.doi:10.1021/ ja8007825
(25)Shen,L.J.;Luo,M.B.;Liu,Y.H.;Liang,R.W.;Jing,F.F.; Wu,L.Appl.Catal.B:Environ.2015,166-167,445.
(26)Chen,F.J.;Cao,Y.L.;Jia,D.Z.;Liu,A.J.Dyes Pigments 2015,120,8.doi:10.1016/j.dyepig.2015.03.030
(27)Chen,F.J.;Cao,Y.L.;Jia,D.Z.Ceram.Int.2015,41, 6645.doi:10.1016/j.ceramint.2015.01.111
(28)Hu,S.Z.;Li,F.Y.;Fan,Z.P.;Wang,F.;Zhao,Y.F.;Lv,Z.B.Dalton Trans.2015,44,1084.doi:10.1039/C4DT02658F
(29)Wang,D.S.;Duan,Y.D.;Luo,Q.Z.;Li,X.Y.;Bao,L.L. Desalination 2011,270,174.doi:10.1016/j.desal.2010.11.042
(30)Lu,M.L.;Pei,Z.X.;Weng,S.X.;Feng,W.H.;Fang,Z.B.; Zheng,Z.Y.;Huang,M.L.;Liu,P.Phys.Chem.Chem.Phys. 2014,16,21280.doi:10.1039/C4CP02846E
(31)Liu,L.Y.;Yang,L.;Pu,Y.T.;Xiao,D.Q.;Zhu,J.G.Mater. Lett.2012,66,121.doi:10.1016/j.matlet.2011.08.025
(32)Ge,L.;Han,C.C.;Liu,J.Appl.Catal.B:Environ.2011,108-109,100.
(33)Liu,H.;Jin,Z.T.;Xu,Z.Z.Dalton Trans.2015,44,14368. doi:10.1039/C5DT01364J
(34)Dong,F.;Zhao,Z.W.;Xiong,T.;Ni,Z.L.;Zhang,W.D.;Sun, Y.J.;Ho,W.K.ACSAppl.Mater.Interf.2013,5,11392.doi: 10.1021/am403653a
(35)Cao,J.;Luo,B.D.;Lin.H.L.;Xu,B.Y.;Chen,S.F. J.Hazard.Mater.2012,217-218,107.
(36)Wang,Y.;Wang,X.C.;Antonietti,M.Angew.Chem.Int.Edit. 2012,51,68.doi:10.1002/anie.201101182
(37)Ge,L.;Han,C.Appl.Catal.B:Environ.2012,117-118,268.
(38)Ma,D.K.;Zhou,H.Y.;Zhang,J.H.;Qian,Y.T.Mater.Chem. Phys.2008,111,391.doi:10.1016/j.matchemphys.2008.04.035
(39)Zhu,Y.P.;Li,J.;Ma,T.Y.;Liu,Y.P.;Du,G.H.;Yuan,Z.Y. J.Mater.Chem.A 2014,2,1093.doi:10.1039/C3TA13636A
(40)Zhang,K.;Kim,W.J.;Ma,M.;Shi,X.J.;Park,J.H.J.Mater. Chem.A 2015,3,4803.doi:10.1039/C4TA05571C
(41)Li,Y.G.;Wei,X.L.;Li,H.J.;Wang,R.R.;Feng,J.;Yun,H.; Zhou,A.N.RSCAdv.2015,5,14074doi:10.1039/ C4RA 14690E
(42)Xu,Y.;Xu,H.;Wang,L.;Yan,J.;Li,H.;Song,Y.;Huang,L.; Cai,G.Dalton Trans.2013,42,7604.doi:10.1039/c3dt32871f
(43)He,B.L.;Dong,B.;Li,H.L.Electrochem.Commun.2007,9, 425.doi:10.1016/j.elecom.2006.10.008
(44)Zhang,J.;Wang,Y.J.;Hu,S.Z.Acta Phys.-Chim.Sin.2015, 31,159.[张健,王彥娟,胡绍争.物理化学学报,2015,31, 159.]doi:10.3866/PKU.WHXB201411201
Preparation of Ternary Metal Sulfide/g-C3N4Hetero junction Catalysts and Their Photocatalytic Activity under Visible Light
WANG Yan-Juan SUN Jia-Yao FENG Rui-Jiang ZHANG Jian*
(Division ofChemistry,ChemicalEngineering and Environment,Liaoning Shihua University, Fushun 113001,Liaoning Province,P.R.China)
A nove l Zn-Mo-CdS/g-C3N4hetero junction photocatalystwas prepared by hyd rothermal posttreatmen tusing dicyandiam ide,zinc acetate,ammonium molybdate,cadm ium acetate,and sodium sulfide as raw materials.X-ray diffraction(XRD),u ltraviolet-visible(UV-Vis),inductive ly coup led plasma atom ic em ission (ICP-AES),electrochem icalimpedance spectroscopy(EIS),and X-ray photoelectron spectroscopy(XPS)were used to characterize the prepared catalysts.The resu lts indicate thathetero junctions are formed across the g-C3N4/Zn-Mo-CdS interface,which promotes interfacialcharge transferand inhibits the recombination ofelectrons and ho les.The ac tivities o f as-p repared ca talysts w ere tested th rough the photoca talytic degrada tion of Rhodamine B(RhB)undervisible light.The results show that the Zn-Mo-CdS/g-C3N4heterojunction photocatalyst clearly displayed increased activity com pared w ith sing le g-C3N4and Zn-Mo-CdS.Atan op timalg-C3N4mass fraction of20%,the as-prepared hetero junction photocatalystdisplayed the highest rate constantunder visible light,which was 30and 10times ofsingle g-C3N4and Zn-Mo-CdS,respectively.Notonly Zn-Mo-CdS,butalsoMo-Ni-CdS and Ni-Sn-CdS can form hetero junctionsw ith g-C3N4to promote the rate ofseparation o felectrons and holes and im prove pho tocata lytic ac tivity.
September 15,2015;Revised:November24,2015;Published onWeb:November30,2015.
Ca rbon nitride;Ternaryme talsulfide;Hetero junction;Pho tocata lysis; Organicmatter degradation
O641;O649
10.3866/PKU.WHXB201511303
*Corresponding author.Email:zhangjianlshu@163.com;Tel:+86-13470570415.
Theprojectwas supported by the Natural Science Foundation of Liaoning Province,China(2015020590)and PilotProgram of University of Liaoning Innovation and Education Reform,China.
辽宁省自然科学基金(2015020590)及辽宁省普通高等学校创新创业教育改革试点专业建设项目资助©Editorialofficeof Acta Physico-Chim ica Sinica
