一种简单高效的新生鼠海马神经元原代培养方法
2016-09-08谢丽赵树进严华成石磊
谢丽, 赵树进, 严华成,3, 石磊
(1.广州军区广州总医院药剂科,广东广州 510010;2.广州中医药大学,广东广州 510006;3.广州军区疾病预防控制中心,广东广州 510507)
·基础研究·
一种简单高效的新生鼠海马神经元原代培养方法
谢丽1,2,赵树进1,严华成1,3,石磊1
(1.广州军区广州总医院药剂科,广东广州510010;2.广州中医药大学,广东广州510006;3.广州军区疾病预防控制中心,广东广州510507)
【目的】建立一种简单、高效、高纯度的新生小鼠海马神经元的原代培养方法。【方法】取出生24 h内的C57BL/6J新生鼠,分离海马组织,采用胰蛋白酶消化加机械吹打的方法获得单个细胞,用含体积分数1%B27及2%Glutamax-100X、终浓度25 μmol/L Glutamate的Neurobasal-A培养基接种培养,3 d后用去Glutamate成分的上述培养基换液,并加阿糖胞苷作用48 h以抑制胶质细胞增长。于倒置相差显微镜下观察细胞的生长状态,采用微管蛋白相关标志物2(MAP2)及Hoechst 33258免疫荧光染色鉴定神经元纯度。【结果】此培养方法获得的海马神经元生长状态良好,细胞形态从接种时的圆形透亮、无突起,到培养第7天后发展为神经元胞体聚集、树突发达并形成纵横交错的神经网络;经鉴定,神经元的纯度高达97.2%以上,且能稳定存活2~3周。【结论】该方法操作简单、高效,所得神经元纯度高,结果稳定。
海马神经元;原代培养;新生小鼠
海马是大脑边缘系统的一个重要部分,参与记忆、学习、认知、情绪等多种高级精神活动[1]。体外培养海马神经元是目前研究阿尔茨海默病、癫痫、脑卒中等神经系统疾病的重要模型[2]。自1977 年Banker等[3]首次在体外成功分离并培养出海马神经元以来,海马神经元的体外分离培养技术一直在不断地发展和完善。但是海马神经元培养过程复杂以及培养条件苛刻,因此如何得到纯度和产量高、活性好的海马神经元仍然值得探讨。胎鼠和新生鼠都可用于海马神经元的培养,但新生鼠的海马神经元的分化程度较胎鼠的高,增加了高纯度培养的难度[4]。本研究通过优化培养条件,尝试获得一种简单、高效、高纯度的新生鼠海马神经元培养方法,为相关中枢神经系统疾病的研究提供稳定的细胞模型,现报道如下:
1 材料和方法
1.1实验动物出生24 h内C57BL/6J小鼠,雌雄不限,普通(SPF)级,由南方医科大学实验动物中心提供,许可证号:SCXK(粤)2011-0015。
1.2主要试剂与仪器胎牛血清(FBS)(以色列Biological Industries公司);多聚赖氨酸(PLL,美国Sigma公司);胰蛋白酶(美国Gibco公司);DMEM/F12(美国 Hyclone公司);DNAse I(瑞士Roche公司);Neurobasal-A培养基(美国Gibco公司,批号:10888022);Glutamax-100X(美国Gibco公司);谷氨酸(美国 Sigma公司);B27(美国Gibco公司);浓缩型正常山羊血清(中国博士德生物公司);微管蛋白相关标志物2(MAP2)抗体(美国Cell Signaling Technology公司);Alexa fluor 488标记的山羊抗兔IgG(美国谷歌生物公司);Hoechst 33258染色液(中国凯基生物公司)。150i/240i二氧化碳恒温培养箱(美国Thermo公司)、Axiovert 40 CFL荧光倒置显微镜(德国蔡司公司)。
1.3海马神经元的培养
1.3.1培养板预处理培养板用工作浓度为0.1 mg/mL的多聚赖氨酸提前铺板,放入37°C培养箱中,半小时后回收多聚赖氨酸,放入37°C培养箱中过夜,临用前用磷酸盐缓冲液(PBS)润洗2次,晾干备用。
1.3.2溶液配制终止消化培养基:DMEM/F12培养基加入体积分数10%FBS、体积分数1%双抗(青霉素/链霉素)。维持培养基:Neurobasal-A培养基加入体积分数 2%B27、体积分数 1% Glutamax-100X、体积分数1%双抗。接种培养基:维持培养基加入终浓度25 μmol/L Glutamate。
1.3.3培养方法取出生 24 h内 C57BL/6J小鼠,用体积分数 75%乙醇消毒,取出大脑组织,置入冰冻的 Hank’s平衡盐溶液(HBSS)中分离双侧海马组织,并仔细剔除血管及脑膜。将分离好的海马组织剪碎至约1 mm3的小块,加入约5倍组织体积的 2.5 g/L胰蛋白酶,放入 37°C孵育箱消化10 min,期间摇晃1次;加入约50 μL的DNA解旋酶以及终止消化的培养基(体积分数10% FBS、DMEM/F12、体积分数1%双抗)终止消化。先加入半量的终止消化液,机械轻柔吹打约 15下,静置 2 min,吸走上层细胞液,再加入另一半,重复上述操作,将剩余液制成单细胞悬液。取20 μL细胞液用于计数,其余 1 000 r/min室温离心5 min,弃去上清液;加接种液培养基,尖嘴吸管轻柔吹打分散细胞后,按4×105个/mL的密度接种于预先包被有多聚赖氨酸的培养板内。放入37°C、体积分数 5%的CO2细胞培养箱中培养。接种72 h后,加入含5 μmol/L阿糖胞苷的维持培养基全量换液,以抑制胶质细胞的增殖,作用48 h后用维持培养基全量换液。之后每隔2 d用维持培养基半量换液,选取不同时间点在倒置显微镜下观察神经元生长状态并拍照。
1.4纯度鉴定取培养7 d的新生鼠海马神经元,用 40 g/L的多聚甲醛室温固 30 min,PBS清洗5 min×3次;加入体积分数0.2% Tritonx-100透化 5 min,体积分数 10%浓缩型正常山羊血清室温封闭 30 min;加入 MAP2抗体(稀释比例为1∶200),4°C冰箱过夜。次日取出,弃一抗,PBS清洗5 min×3次。避光加入Alexa Fluor 488标记的二抗(稀释比例为1∶100),室温30 min后,PBS清洗 5 min×3次;加入Hoechst 33258染色液,避光染色 5~10 min,PBS清洗 5 min×3次;加入PBS,在倒置荧光显微镜下观察,拍照,记录,用Adobe PhotoShop CS6软件制作MAP2染色图与Hoechst 33258染色图的叠加图,并计算纯度百分比。根据带绿色荧光的细胞占总细胞的比例即可判断神经元的纯度。
2 结果
2.1形态学观察图1结果显示:培养1 d后细胞贴壁良好,呈圆形、椭圆形或锥体形,立体感强,部分细胞已伸出短小突起;培养第3天,胞体饱满,折光性与立体感强,突起继续增长,可见单极、双极、多极多种突起,有些突起相互连接;培养第5天时,神经元胞体开始聚集,突起增多并相互连接呈网络;培养第 7天,神经元树突发达,神经元突起更密集,纵横交错,难以辨认突起的起源,形成发达的神经网络。
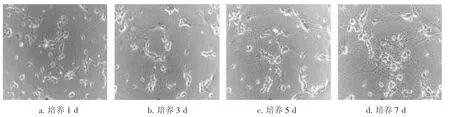
图1 不同培养时间海马神经元的形态(×200)Figure 1 Histology of mouse hippocampal neurons at different culturing time(×200)
2.2神经元免疫荧光鉴定及纯度鉴定图2结果显示:培养至7 d的海马神经元细胞形态比较典型,胞体饱满,轴突、树突粗大,相互交错形成神经网络,此时利用神经元特异性标志物微管蛋白相关标志物2(MAP2)免疫荧光染色来鉴定神经元及检验其纯度。MAP2阳性细胞胞体和树突呈绿色,轴突及神经胶质细胞均不染色,同时采用Hoechst 33258 DNA荧光素对海马神经元细胞核进行蓝染获得总细胞数量,正常海马神经元细胞核光滑,呈卵圆形,亮度均一。叠加图中胞体染细胞和核染细胞基本上都能重合。计算MAP2阳性细胞占总细胞的百分率,神经元的纯度达97.2%。
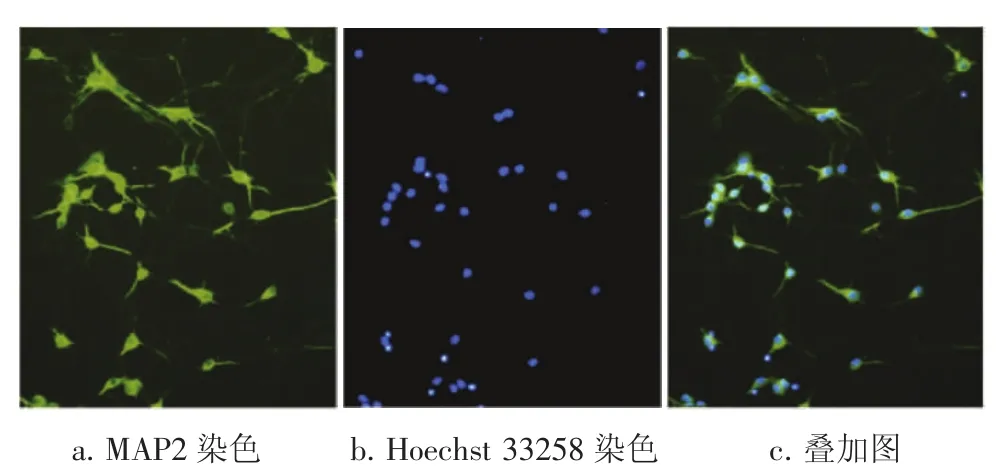
图2 神经元的鉴定(×200)Figure 2 Identification of mouse hippocampal neurons(×200)
3 讨论
海马神经元原代培养是研究神经系统结构及功能的重要技术之一。由于海马取材过程中难以完全分离神经元与胶质细胞,而胶质细胞可增殖从而影响后期神经元的实验研究;再加上操作技术复杂难于掌握,因此要稳定获得纯度高、活性好的海马神经元仍然很不容易。目前国内外根据使用培养液的不同可分为多种培养方法[5]。以往神经元的原代培养一般采用DMEM培养基,并用阿糖胞苷来抑制神经胶质细胞的增长[6]。但因为DMEM培养基也非常适合其他杂质细胞的生长,而阿糖胞苷的用量过大亦会对神经元生长造成影响,故此种方法获得的海马神经元纯度不高。同时有改进的方法采用DMEM/F12培养液作为种植液,4 h后用含B27的Neurobasal-A 培养基换液继续培养[7-8],但细胞培养4 h后细胞贴壁不牢固,换液会增加神经元的损失。
本研究中Neurobasal-A培养基(10888022)是美国Gibco公司生产的专门用于新生鼠神经元培养的培养基;B27添加剂中含有多种神经细胞生长的必需神经因子,因此添加B27而非血清可促进神经元的生长而抑制非神经元的生长和繁殖[9];Glutamine对神经元有保护作用,但此溶液不稳定,每次用时需现配现加,而Glutamax-100X能在培养中长期稳定地给神经元提供代谢所需的Glutamine,从而有利于神经元的长期存活[10];Glutamate参与突触的形成和维持[11],在接种后的前3 d,培养基中加入Glutamate可加快神经元突触生长,促使神经元更好地贴壁,提高存活率。本研究选择的接种液培养基结合以上成分优点,为新生鼠海马神经元提供了一个良好的生长环境,24 h后已有一部分神经元突触明显。3 d后换用去Glutamate的维持培养基并加入0.1 mg/mL阿糖胞苷,进一步抑制非神经元细胞(主要是胶质细胞)的增长。为了避免阿糖胞苷对神经元的影响,48 h后用维持培养基全量换液。由此方法培养的新生鼠海马神经元7 d后可形成纵横交错的神经网络,用MAP2免疫荧光染色法对神经元进行鉴定,纯度可达97.2%。
由于海马神经元取材过程的复杂性以及其本身细胞的脆弱性,因此在进行培养的实际操作中需反复试验摸索,步步细心谨慎。关键步骤中有几点值得注意:①为避免多聚赖氨酸多神经元的影响,包被回收后需用PBS清洗2遍。②保证分离提取海马的整个过程在冰台低温环境中进行,所用到的HBSS也应预冷。③在取脑过程中尽量避免血液的残留,可用预冷的HBSS冲洗2次。在剥离血管和脑膜时应彻底剥离干净。④胰酶消化的时间不宜过长也不宜过短,一般10 min左右即可。⑤终止消化吹打获得单细胞悬液时应动作轻柔,本研究采用分步吹打的方法,一次吹打15~20下后将上层细胞吸走,既避免了反复吹打的机械损伤,又能充分分离成单个细胞。无需过筛,减少了过筛过程的细胞损失。⑥细胞密度对神经元的生长尤为重要,密度过低导致神经元不能相互接触,影响细胞的存活与成熟,密度过高亦可致接触抑制,以及细胞聚集成团。因此培养时需认真计数,细胞密度为4×105~5×105个/mL为宜。⑦在接种后的12 h内尽量避免晃动细胞板,因为此时间段有些神经元还未贴壁或贴壁不牢,以防细胞的聚集,导致细胞分布不均匀。⑧加入阿糖胞苷处理后,需全量换液,之后采用半量换液的方式,因为神经元自身会分泌神经营养因子类物质,换半液有助于保留这些营养物质。
综上所述,本研究所用的新生鼠海马神经元的培养方法流程简化、操作简单,培养所得的神经元活性高、纯度高,可稳定培养至3周,值得推广运用。
[1]Kim S,Dede A J,Hopkins R O,et al.Memory,scene construction,and the human hippocampus[J].Proc Nat Acad Sci USA,2015,112(15):4767.
[2]黄孝闻,王绪平,寿旦.海马神经元细胞模型及其应用的研究进展[J].中华中医药学刊,2015(11):2730.
[3]Banker G A,Cowan W M.Rat hippocampal neurons in dispersed cell culture[J].Brain Res,1977,126(3):342.
[4]李春莉,杨宝峰,张景海,等.新生大鼠海马和皮层神经元的原代培养及鉴定[J].沈阳药科大学学报,2011(4):299.
[5]徐东芳,朱长太,刘东梅,等.三种培养液进行海马神经元原代培养的对比研究[J].安徽医科大学学报,2006(5):521.
[6]翁启芳.新生大鼠大脑皮层神经元的原代培养[J].中国医药指南,2013(20):87.
[7]张学平,王高华,王惠玲,等.海马神经元原代培养与纯度鉴定方法的优化[J].武汉大学学报(医学版),2013(5):703.
[8]张勇,张国宁,丛敬,等.胎鼠海马神经元原代培养及转染条件的优化[J].解剖科学进展,2011,17(5):464.
[9]Brewer G J,Torricelli J R,Evege E K,et al.Optimized survival of hippocampal neurons in B27-supplemented Neurobasal,a new serum-free medium combination[J].J Neurosci Res,1993,35 (5):567.
[10]Stelmashook E V,Novikova S V,Isaev N K.Glutamine effect on cultured granule neuron death induced by glucose deprivation and chemical hypoxia[J].Biochemistry(Mosc),2010,75(8):1039.
[11]Levenson J M,Weeber E J,Sweatt J D,et al.Glutamate uptake in synaptic plasticity:from mollusc to mammal[J].Curr Mol Med,2002,2(7):593.
【责任编辑:黄玲】
A Simple and Effective Primary Culture Method for Newborn Mouse Hippocampal Neurons
XIE Li1,2,ZHAO Shujin1,YAN Huacheng1,3,SHI Lei1
(1.Dept.of Pharmacy,Guangzhou General Hospital of Guangzhou Military Command,Guangzhou 510010 Guangdong,China;2.Guangzhou University of Chinese Medicine,Guangzhou 510006 Guangdong,China;3.Center for Disease Prevention and Control,Guangzhou Military Command,Guangzhou 510507 Guangdong,China)
ObjectiveTo establish a simple,effective and high-pureity primary culture method for newborn mouse hippocampal neurons.Methods C57BL/6J newborn mice within 24 hours after birth were used for the isolation of the hippocampus.Mechanical pipetting combined with enzymic digestion with trypsin was used to obtain monoplast suspension,and then the neurons were planted in Neurobasal-A medium containing volume fraction of 1%B27,2% Glutamax-100X and 25 μmol/L of Glutamate.After culturing for 3 days,the initial medium for the neurons was totally replaced by the above medium with Glutamate free and with cytosine arabinoside(Ara-C)added to inhibit the growth of glial cells for 48 hours.The morphological changes of neuron cells were observed under inverted phase-contrast microscope.Microtubule-associated protein-2(MAP2)and Hoechst33258 immunofluorescence were used to identify the purity of neurons.Results The growth of hippocampal neurons obtained from the culture method was well on the 7th day of culturing,showing as neuronal soma aggregation,well-developed cytodendrite and formation of criss-crossed nerve net,which were quite different from the round,transparent,and process-free cells at the beginning of planting.The purity of the obtained neurons was more than 95%and the neurons could survive for 2-3 weeks stably.Conclusion The method is simple and effective for obtaining the high-purity and stable neurons.
hippocampal neurons;primary culture;newborn mice
R34
A
1007-3213(2016)04-0570-04
10.13359/j.cnki.gzxbtcm.2016.04.029
2016-01-14
谢丽(1989-),女,硕士研究生;E-mail:1032070376@qq.com
石磊(1962-),女,教授;E-mail:lucyshi622_921@163.com
广东省科技计划项目(编号:2013A022100035);广州市科技计划项目(编号:201509010012);广州市科技基金项目(编号:2013J2200027)
