甘氨酸二肽分子酰胺-I带光谱与结构相关性
2016-09-06蔡开聪刘亚男留珊红杜芬芬
蔡开聪 郑 轩 刘亚男 留珊红 杜芬芬
(1福建师范大学化学与化工学院,福州350007;2福建省理论与计算化学重点实验室,福建厦门361005)
甘氨酸二肽分子酰胺-I带光谱与结构相关性
蔡开聪1,2,*郑轩1,2刘亚男1,2留珊红1,2杜芬芬1,2
(1福建师范大学化学与化工学院,福州350007;2福建省理论与计算化学重点实验室,福建厦门361005)
系统探索了蛋白质二肽模型分子——甘氨酸二肽(GLYD)在气相与水溶液中的结构与光谱特性。从分子动力学轨迹中提取具有代表性结构的GLYD-D2O聚集体的瞬态结构开展简正模式分析,获取了对蛋白质二级结构敏感的酰胺-I带的振动光谱参数,建立起振动光谱与特征基团结构间的相关性。将溶剂作用以静电势场的形式投影至二肽分子骨架中,与酰胺-I带在气/液相中的频率差相关联,并引入酰胺-I带简正模式随二级结构变化的规律,将各个构象态可能存在的振动耦合包含在内,构建具有二级结构敏感性的静电频率转换图,实现溶液相中多肽骨架酰胺-I带的快速准确预测。
甘氨酸二肽;酰胺-I带;振动光谱;静电频率图
蛋白质及多肽的骨架上酰胺-I带(主要为C=O的伸缩振动)在中红外区域具有强烈的振动吸收,其特征吸收峰对分子骨架二级结构的变化十分敏感,因此广泛用作蛋白质及多肽的结构探针5。酰胺-I带的特征振动吸收谱带位于1600-1700 cm-1,对于特定的二级结构可以用“指纹”识别的方法进行鉴定:如α螺旋在1655 cm-1处有一个特征吸收峰,且随着螺旋链的增长而红移;而β折叠则出现明显的双峰,其在1630 cm-1处有个较强吸收峰,在1685 cm-1处吸收峰相对较弱6-9。蛋白质骨架酰胺吸收带的零级频率预测一直是一个难题,由于酰胺单元的跃迁偶极矩之间存在振动耦合作用,其振动光谱表现为一个较宽的吸收带,使得传统的一维红外光谱学手段难以有效地将各个组分区分开。为此,人们常常引入同位素取代的方法,如将酰胺-I带中C=O基团修饰为13C=18O,从而使其频率发生约60 cm-1的红移,剥离出酰胺-I带的光谱区域,从而实现特定位点光谱和结构相关性解析10,11。
许多重要的生命过程都是在溶液环境下进行的,要发展蛋白质结构检测方法,需要在微观层面上描述蛋白质体系在溶液相中的动态结构以及包括溶剂在内的微环境。飞秒激光二维红外光谱等实验新技术的发展使得人们在更小更短暂的时空领域能够观测到包含微观世界中蛋白质结构涨落信息的实验信号12-18,而这些实验信号需要在理论层面上发展新方法来深入认识和解析溶液中蛋白质结构及相应的酰胺振动吸收带的光谱特性。在水溶液中,溶质-溶剂间的静电相互作用占据了主导地位,因此人们提出了用于实现酰胺-I带振动频率快速预测的静电频率转换图9,19-26。该模型将溶剂作用以静电势场的形式投影至多肽骨架中,与酰胺-I带在气相和液相中的频率差相关联,通过分子所处的静电势场环境,实现酰胺-I带振动光谱的快速准确预测。静电频率转换图的构建主要基于模型分子-氮甲基乙酰胺(NMA)以及非天然氨基酸模型分子(NEPA)27-29。尽管多肽骨架对酰胺-I带光谱频率具有显著的影响,针对具有二级结构的多肽乃至寡聚肽所构建的模型仍少有报道,因为要考察特定结构多肽的酰胺吸收带之间的振动耦合,需要通过较为复杂的跃迁偶极耦合作用才能有效表征多肽链中酰胺-I带的光谱特性21,23,26,30,更为经验和简化的方法有待开发。
本文开展分子动力学模拟,获取了甘氨酸二肽(GLYD)在水溶液中的全原子运动轨迹,探究其动态结构及存在的溶质-溶剂相互作用。从动力学瞬态结构中提取第一溶剂化层中具有代表性的溶质-溶剂聚集体,开展简正模式分析,了解酰胺-I带振动光谱参数及溶剂对其所产生的影响。探索气相中酰胺-I带频率的二级结构依赖性,了解各个构象态下两个酰胺-I带的特征简正振动频率,将各个构象态中可能存在的振动耦合考虑在内。进一步将溶剂作用以静电势场的形式投影至酰胺单元上,辅以酰胺-I带简正模式的二级结构依赖性,从而构建静电频率转换模型,实现多肽分子骨架酰胺-I带光谱的快速准确预测。
2 计算方法
2.1分子动力学模拟
借助NAMD软件31开展全原子分子动力学模拟,探索GLYD在重水中的结构动力学特性。GLYD采用CHARMM力场(版本号:c35b2)描述32,水溶液则采用TIP3P模型描述33。研究体系为1个GLYD分子和2279个重水分子组成的4.2 nm的立方体盒子。体系设置了周期性边界环境,长程静电作用采用了particle mesh Ewald(PME)方法计算,非键相互作用的截断距离设定为1.2 nm。
体系采用共轭梯度法进行了10000步的能量最小化,排除了可能的高能量和空间重叠构型,随后逐步升温至室温(298 K)。在恒温恒压系综(NPT)下采用Nosé-Hoover Langevin piston方法进行了分子动力学模拟,在298 K温度下以5 fs的步长采集了1 ns全原子运动轨迹。
2.2量子化学计算
从1 ns的全原子运动轨迹中等间隔提取了其中20个瞬态结构,对这20个结构分别提取含有重水个数n=1-5的GLYD-nD2O聚集体(合计100个)开展量子化学计算,并在GLYD-5D2O聚集体外添加连续极化介质模型(PCM)补偿体相中水的影响。在B3LYP/6-31+G(d)水平上对这120个聚集体进行结构优化和简正模式分析,同时借助势能分布分析(PED)方法34,对酰胺-I带进行指认。
在B3LYP/6-31+G(d)水平上对气相中GLYD的骨架二面角(Φ:∠CNCC;Ψ:∠NCCN)进行扫描,固定其中一个二面角,对另一个二面角进行旋转(步长为10°)。对得到的1369个构象异构体进行几何结构优化和简正模式分析,获取各个构象异构体的酰胺-I带的振动频率,考察其与多肽二级结构之间的相关性。所有的量子化学计算均在Gaussian 09软件35下进行。
2.3静电频率转换图的构建
将经过量子化学计算优化的GLYD-nD2O聚集体(共120个)作为建模样本。选取的样本具有结构代表性,又具有电子结构准确性,成键和非键相互作用已经通过分子动力学模拟和量子化学计算隐性地在样本结构特征中表现出来。将其中溶剂原子看作质点,GLYD骨架上甲基、亚甲基看作联合原子(电荷集中在中心原子上),计算其在酰胺单元的原子位点(C、O、N、H)上所产生的静电势场,同时引入酰胺-I带简正模式随骨架二面角变化的特征,将其与GLYD中酰胺-I带的气/液相频移相关联:

其中,vl为GLYD-nD2O聚集体中酰胺-I带的频率值(均乘以校正因子0.974,即气相实验值vg与对应的C5构象下在B3LYP/6-31+G(d)水平上计算频率值之比的均值)36,vg为GLYD处于C5构象时酰胺-I带的气相实验频率值(1693 cm-1,酰胺-Ia带;1707 cm-1,酰胺-Ib带)37,f为模型参数,φ为酰胺单元上所产生的静电势,ω为处于不同构象态时GLYD中酰胺-I带频率与气相值vg的差值。通过求解超定方程组,获得静电频率图转换参数(表1)。

表1 甘氨酸二肽中酰胺-I带的静电频率转换图参数Table 1 Parameters of the electrostatic frequency map for the amide-I band of GLYD
将模型参数应用于全原子动力学轨迹中,得到修正后的酰胺-I带频率轨迹及其分布态密度(DOS),并通过如下线型函数计算得到振动吸收光谱图,

其中,I(v)为光谱强度,v为随时间变化的酰胺-I带的瞬时振动频率,t为时间,,为酰胺-I带频率的平均值,T1为酰胺-I带第一激发态的寿命(0.5 ps)38。
3 结果与讨论
3.1水溶液中GLYD的微观结构
借助空间分布函数(SDF)和径向分布函数(RDF),系统考察GLYD的酰胺单元与水分子之间可能存在的氢键相互作用及其强弱关系(图1)。从空间分布函数(图1C)中可以看出,GLYD两个酰胺单元上的C=O在较短的距离范围内均被蓝色的区域所包围,表明C=O上的O原子与水的D原子形成较强的氢键作用;而N―H周围则被红色的区域包围,表明N―H中的H原子倾向于和水中的O原子结合形成氢键。
通过径向分布函数(图1(A,B))可以看出,两个酰胺单元上的C=O和N―H基团分别与重水中的D和O原子形成较强的氢键作用。其中,C=O中的O原子与重水中的D原子在距离分别为0.175 nm处均形成较强的峰(g(r)a=1.21;g(r)b=1.25);而N―H的H原子与重水中的O原子在距离分别为(a) 0.195 nm和(b)0.205 nm处形成两个峰(g(r)a= 0.80;g(r)b=0.69)。通过对第一水合层内的g(r)进行积分,可以得到第一溶剂化层内的溶剂配位数(N),

其中,ρ为密度,Rmin为g(r)第一个最小值出现的距离,r为原子间距离。研究结果表明,GLYD的两个酰胺单元的C=O基团第一溶剂化层中围绕着两个D2O分子,N―H基团周围围绕着一个D2O分子。由于酰胺单元的亲水性及氢键作用,GLYD在重水溶液中具有良好的溶解能力。
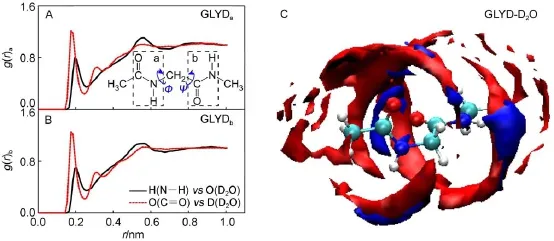
图1 甘氨酸二肽与水分子形成的径向分布函数(A,B)与空间分布函数(C)Fig.1 Radial distribution function(A,B)and spatial distribution function(C)between GLYD and D2OMolecular structure of GLYD is shown in Fig.A,and the amide units are denoted as“a”and“b”.In Fig.C,blue:water hydrogen;red:water oxygen. color online
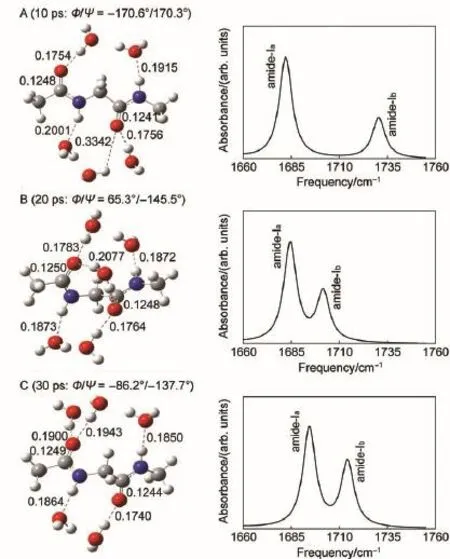
图2 甘氨酸二肽与周围重水分子的瞬态结构及相应的酰胺-I带的计算光谱Fig.2 Instantaneous structures of GLYD-D2O clusters and corresponding calculated amide-I spectra bond length in nm
3.2酰胺-I带光谱与结构相关性
我们选取了在10、20和30 ps时刻,GLYD与周围D2O(水分子数n=5)所形成的聚集体的瞬态结构,在密度泛函理论水平上计算得到了3N-6个振动模式的振动频率。其中,处于1660-1760 cm-1的酰胺-I带的振动吸收峰与分子结构具有显著的相关性(图2)。由于酰胺单元中两个C=O基团倾向于和重水形成较强的氢键作用,C=O双键的键长依据氢键强弱的不同发生一定程度的拉伸,并在酰胺-I带的光谱区域产生相应的频率红移。
在动力学轨迹第10 ps的瞬态时刻(图2A),乙酰端(C=O,0.1248 nm)a的键长大于氨基端(C=O,0.1241 nm)b,对应的酰胺-Ia带的振动频率相比酰胺-Ib带红移了48.4 cm-1;在20 ps的瞬态时刻(图2B),(C=O,0.1248 nm)b由于和周边的水分子同时形成两个较强的氢键,酰胺-Ib带振动频率红移至1701.7 cm-1,与酰胺-Ia带的频率差缩小至17.2 cm-1;在30 ps的瞬态时刻(图2C),溶质-溶剂间氢键作用较弱,两个C=O双键键长较20 ps时有所缩短,酰胺-I带频率整体蓝移。随着时间的迁移,GLYD及周围重水处于不停的热运动之中,溶质-溶剂相互作用对GLYD二级结构的影响在酰胺-I带中表现出来,使得光谱表象成为有力的结构探测信号。
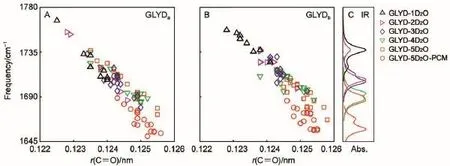
图3 酰胺-I带频率与C=O键长的相关性(A,B)及第140 ps瞬态时刻GLYD-nD2O的酰胺-I带IR光谱(C)Fig.3 Correlation between the amide-I frequencies and the C=O bond lengths(A,B),and the calculated IR spectra of amide-I mode for selected GLYD-nD2O clusters at 140 ps(C)Abs.:normalized absorbance.color online
对等间隔选取的GLYD-nD2O(n=1-5)聚集体开展量子化学计算得到优化后的结构及对应的酰胺-I带振动频率,其相关性如图3所示。在140 ps瞬态时刻,随着GLYD周围重水分子个数的增加,C=O键长由于氢键的作用而拉伸变长,导致相应的酰胺-I带的振动频率发生的红移程度加剧(图3C),C=O可能同时和两个重水分子分别形成分子间氢键。对120个聚集体的酰胺-I带进行统计分析,发现酰胺-Ia带频率均值分布从1717.9 cm-1(GLYD-1D2O)至1703.5 cm-1(GLYD-5D2O),酰胺-Ib带频率均值分布从1741.3 cm-1(1D2O)至1695.9 cm-1(5D2O)。在GLYD-5D2O聚集体外层添加了PCM模型补偿溶液体相作用后,酰胺-I带振动频率仍有一定程度的红移,频率均值分别红移至1664.5 cm-1(酰胺-Ia带)和1668.5 cm-1(酰胺-Ib带)。溶剂产生的酰胺-I带频率红移主要来自第一水合层内重水所产生的氢键作用,但是来自体相的作用不可忽视。
酰胺-I带光谱的频移除了受到溶质-溶剂间氢键作用的影响,GLYD本身二级结构的构象态变化也会导致光谱吸收带的迁移。通过对气相中遍及整个拉式构象图的孤立的GLYD构象异构体进行简正模式分析,并借助PED分析方法对酰胺-I带进行了系统的指认和归属,得到了气相中GLYD分子两个酰胺-I带的振动频率随分子二级结构变化的规律(图4)。
酰胺-Ia/-Ib带频率在拉式图中沿反对角线呈对称分布。其中,酰胺-Ia带的频率分布范围从1722.3 cm-1(Φ/Ψ=80°/-70°)至1824.0 cm-1(Φ/Ψ= 0°/180°),酰胺-Ib带的频率则分布在1720.4 cm-1(Φ/ Ψ=-10°/10°)至1796.1 cm-1(Φ/Ψ=-50°/-90°)。酰胺-Ia带的振动频率平均值(1760.6 cm-1)略高于酰胺-Ib带(1758.0 cm-1)。简正模式分析结果揭示了GLYD各个构象异构体均有特异性的酰胺-I带指纹吸收频率,且简正模式包含了可能存在的振动耦合、费米共振等影响光谱频移的因素。如此构建的构象异构体的酰胺-I带频率数据库能够有效地将二级结构变化以光谱表象的形式进行表达。
在气相或是溶液中,分子的热运动导致GLYD的结构涨落遵循一定的规律,分子倾向于形成较为稳定的构象。在气相中,孤立的GLYD的两个酰胺单元倾向于形成分子内氢键,使得分子呈现C5和C7构象39。而在水溶液中,酰胺单元和水分子间存在较强的氢键作用,分子动力学模拟结果表明GLYD倾向于形成PPII,β-折叠,α-螺旋等构象。由于绝大多数生命活动都是在溶液环境下发生的,要考虑溶液相中溶剂作用对光谱的影响,则需要将极性溶剂环境下占主导地位的静电作用进行量化。同时,溶剂作用使得GLYD构象态发生改变,溶剂作用隐性地在结构中发生作用,此时将上述简正模式分析中构象态对光谱参数的影响引入静电频率转换图中(公式(1),ω(Φ,Ψ)),使其具有二级结构的敏感性,结合溶剂中占主导地位的静电作用,与气/液相中酰胺-I带频移相关联,从而计算获取转换图参数。借助模型方法,只需知道多肽所呈现的折叠特性及溶剂分子在酰胺单元上所产生的静电势场作用,就能够有效实现多肽乃至蛋白质特定位点酰胺-I带振动吸收峰的快速准确预测。
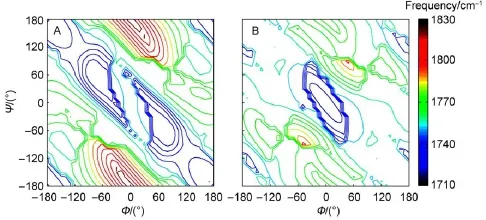
图4 酰胺-I带振动频率随GLYD骨架二面角(Φ/Ψ)变化分布Fig.4 Amide-I frequency distributions due to the backbone dihedrals(Φ/Ψ)of GLYD(A)amide-Ia;(B)amide-Ib.color online
3.3酰胺-I带光谱模拟
计算整个动力学轨迹中溶剂和GLYD骨架在酰胺单元上所产生的静电势场作用,结合气相中酰胺-I带的二级结构依赖性,将模型参数(表1)应用于整个分子动力学轨迹中,得到优化后的GLYD中酰胺-I带的频率统计分布(图5,DOS),进一步通过线型函数(方程(2))可以计算得到其红外吸收光谱(图5,IR)。
经过模型优化得到两个酰胺-I带的最可几频率分别位于1631 cm-1(酰胺-Ia)和1646 cm-1(酰胺-Ib),其统计分布均呈现高斯型分布,拟合得到的半高全宽(FWHM)分别为34.1 cm-1(酰胺-Ia)和34.0 cm-1(酰胺-Ib),且均位于实验光谱吸收带的范围内。由于GLYD的Cα上的R基仅有H原子,分子具有一定的对称性,因此水溶液中GLYD的FTIR光谱仅呈现出一个振动吸收峰(图5)40。为了便于和实验值比较,将两个酰胺-I带的频率轨迹进行了叠加之后进行统计分析(图5,Sum-DOS),其最可几分布值(1639.0 cm-1)较好地重现了实验光谱的吸收峰值(1640.3 cm-1),表明实验测试得到的GLYD光谱虽然表现为单峰,但实际上是由两个十分接近的组分构成,两个吸收峰对应的化学结构有所不同。

图5 重水溶液中GLYD两个酰胺-I带频率的统计分布及模拟红外光谱Fig.5 Static frequency distributions and the simulated IR spectra for the amide-Iaand-Ibmodes of GLYD in D2OThe experimental spectrum is taken from Cormanich et al.40. DOS:density of state;Sum-DOS:sum of density of state. color online
然而DOS并不是真实的红外光谱,由于未考虑到运动窄化作用41,42,酰胺-I带的谱带宽度(Sum-DOS,37.7 cm-1)大于其实验观测值(33.5 cm-1)。通过线型函数对两个酰胺-I带分别进行了IR模拟,所得到的局域模式(图5)的吸收峰频率和统计分布的最可几频率一致,其FWHM相较对应的DOS有一定程度的窄化,分别为22.2 cm-1(酰胺-Ia)和23.6 cm-1(酰胺-Ib)。同时,考虑到两个酰胺-I带之间的振动耦合,我们还采用此耦合方法进行了光谱的模拟43,然而该方法并不适用于现有模型得到的频率轨迹,得到的红外光谱具有明显的双峰特性(数据未给出)。此外,在构建模型时已经引入了酰胺-I带简正模式的二级结构依赖性,在一定程度上包含了振动模式之间的耦合作用。模型的建立能够有效实现光谱参数的快速预测,借助模型参数获得光谱数据的计算量远远小于量子化学计算,同时不受到样本大小、计算方法、基组水平的影响,并能够帮助人们解析实验光谱信号在真实世界中所表达的物理化学意义,进而依据光谱表象,有效描述溶液中多肽的结构特性及所处化学微环境特征。
4 结论
溶液相中蛋白质和多肽空间结构的解析一直是一个研究热点。本文结合量子化学计算方法和分子动力学模拟手段,获取了飞秒至纳秒时间尺度下甘氨酸二肽的结构动力学特性。通过空间分布函数和径向分布函数,对可能存在的溶质-溶剂相互作用有了系统的认识。提取了分子动力学轨迹中不同瞬态时刻分子聚集体开展量子化学计算,了解溶质-溶剂间相互作用对多肽二级结构的影响及其相关的光谱表象,进一步等间隔选取了分子动力学轨迹中具有代表性结构的GLYD-nD2O聚集体,探索溶剂作用对酰胺-I带光谱频移的影响。考察气相中孤立GLYD各个构象异构体的酰胺-I带的简正振动频率,认识光谱表象的二级结构依赖性及其潜在的振动耦合等作用。建立起溶剂静电作用、酰胺-I带简正模式随构象态变化规律与酰胺-I带在气/液相中光谱频移之间的相关性,构造具有二级结构敏感性的静电频率转换模型,实现溶液相中对多肽二级结构敏感的酰胺-I带光谱参数的快速准确预测。
致谢:感谢中国科学院超算中心网格计算提供的高性能计算服务。
References
(1)Carrell,R.W.;Lomas,D.A.Lancet 1997,350,134.doi: 10.1016/S0140-6736(97)02073-4
(2)Savelieff,M.G.;DeToma,A.S.;Derrick,J.S.;Lim,M.H. Accoutns Chem.Res.2014,47,2475.doi:10.1021/ar500152x
(3)Dill,K.A.;MacCallum,J.L.Science 2012,338,1042.doi: 10.1126/science.1219021
(4)DeToma,A.S.;Salamekh,S.;Ramamoorthy,A.;Lim,M.H. Chem.Soc.Rev.2012,41,608.doi:10.1039/C1CS15112F
(5)Krimm,S.;Bandekar,J.Adv.Protein Chem.1986,38,181.
doi:10.1016/S0065-3233(08)60528-8
(6)Barber-Armstrong,W.;Donaldson,T.;Wijesooriya,H.;Silva, R.A.G.D.;Decatur,S.M.J.Am.Chem.Soc.2004,126, 2339.doi:10.1021/ja037863n
(7)Huang,C.Y.;Getahun,Z.;Zhu,Y.;Klemke,J.W.;DeGrado, W.F.;Gai,F.Proc.Natl.Acad.Sci.U.S.A.2002,99,2788. doi:10.1073/pnas.052700099
(8)Du,D.;Zhu,Y.;Huang,C.Y.;Gai,F.Proc.Natl.Acad.Sci.U. S.A.2004,101,15915.doi:10.1073/pnas.0405904101
(9)Malolepsza,E.;Straub,J.E.J.Phys.Chem.B 2014,118, 7848.doi:10.1021/jp412827s
(10)Woys,A.M.;Almeida,A.M.;Wang,L.;Chiu,C.C.; McGovern,M.;de Pablo,J.J.;Skinner,J.L.;Gellman,S.H.; Zanni,M.T.J.Am.Chem.Soc.2012,134,19118.doi: 10.1021/ja3074962
(11)Kim,Y.S.;Wang,J.;Hochstrasser,R.M.J.Phys.Chem.B 2005,109,7511.doi:10.1021/jp044989d
(12)Moran,S.D.;Zanni,M.T.J.Phys.Chem.Lett.2014,5,1984.
doi:10.1021/jz500794d
(13)Jones,K.C.;Peng,C.S.;Tokmakoff,A.Proc.Natl.Acad.Sci. U.S.A.2013,110,2828.doi:10.1073/pnas.1211968110
(14)Kim,H.;Cho,M.Chem.Rev.2013,113,5817.doi:10.1021/ cr3005185
(15)Tucker,M.J.;Abdo,M.;Courter,J.R.;Chen,J.;Brown,S.P.; Smith,A.B.;Hochstrasser,R.M.Proc.Natl.Acad.Sci.U.S. A.2013,110,17314.doi:10.1073/pnas.1311876110
(16)Kim,Y.S.;Hochstrasser,R.M.J.Phys.Chem.B 2009,113, 8231.doi:10.1021/jp8113978
(17)Wang,J.P.Chin.Sci.Bull.2007,52,1221.[王建平.科学通报,2007,52,1221.]
(18)Zheng,J.R.Physics 2010,39,162.[郑俊荣.物理,2010,39, 162.]
(19)Carr,J.K.;Zabuga,A.V.;Roy,S.;Rizzo,T.R.;Skinner,J.L. J.Chem.Phys.2014,140,224111.doi:10.1063/1.4882059
(20)Jansen,T.L.C.J.Phys.Chem.B 2014,118,8162. doi:10.1021/jp5012445
(21)Reppert,M.;Tokmakoff,A.J.Chem.Phys.2013,138,134116/ 1.doi:10.1063/1.4798938
(22)Lin,Y.S.;Shorb,J.M.;Mukherjee,P.;Zanni,M.T.;Skinner, J.L.J.Phys.Chem.B 2009,113,592.doi:10.1021/jp807528q (23)Wang,L.;Middleton,C.T.;Zanni,M.T.;Skinner,J.L. J.Phys.Chem.B 2011,115,3713.doi:10.1021/jp200745r
(24)Dijkstra,A.G.;Jansen,T.L.C.;Knoester,J.J.Phys.Chem.B 2011,115,5392.doi:10.1021/jp109431a
(25)Lee,H.;Choi,J.H.;Cho,M.J.Chem.Phys.2012,137, 114307.doi:10.1063/1.4751477
(26)Reppert,M.;Tokmakoff,A.J.Chem.Phys.2015,143,061102.doi:10.1063/1.4928637
(27)Cai,K.;Su,T.;Lin,S.;Zheng,R.Spectrochim.Acta A 2014, 117,548.doi:10.1016/j.saa.2013.08.058
(28)Shi,J.P.;Zhao,J.;Yang,F.;Wang,J.P.Acta Phys.-Chim.Sin. 2013,29,695.[石纪培,赵娟,杨帆,王建平.物理化学学报,2013,29,695.]doi:10.3866/PKU.WHXB201302213
(29)Cai,K.;Du,F.;Zheng,X.;Liu,J.;Zheng,R.;Zhao,J.;Wang, J.J.Phys.Chem.B 2016,120,1069.doi:10.1021/acs. jpcb.5b11643
(30)Jansen,T.L.C.;Knoester,J.J.Phys.Chem.B 2006,110, 22910.doi:10.1021/jp064795t
(31)Phillips,J.C.;Braun,R.;Wang,W.;Gumbart,J.;Tajkhorshid, E.;Villa,E.;Chipot,C.;Skeel,R.D.;Kale,L.;Klaus,S. J.Comput.Chem.2005,26,1781.doi:10.1002/jcc.20289
(32)MacKerell,A.D.,Jr.;Bashford,D.;Bellott,M.;Dunbrack,R. L.,Jr.;Evanseck,J.D.;Field,M.J.;Fischer,S.;Gao,J.;Guo, H.;Ha,S.;Joseph-McCarthy,D.;Kuchnir,L.;Kuczera,K.; Lau,F.T.K.;Mattos,C.;Michnick,S.;Ngo,T.;Nguyen,D. T.;Prodhom,B.;Reiher,W.E.,III;Roux,B.;Schlenkrich,M.; Smith,J.C.;Stote,R.;Straub,J.;Watanabe,M.;Wiorkiewicz-Kuczera,J.;Yin,D.;Karplus,M.J.Phys.Chem.B 1998,102, 3586.doi:10.1021/jp973084f
(33)Jorgensen,W.L.;Chandrasekhar,J.;Madura,J.D.;Impey,R. W.;Klein,M.L.J.Chem.Phys.1983,79,926.doi:10.1063/ 1.445869
(34)Jamróz,M.H.Vibrational Energy Distribution Analysis VEDA 4;Warsaw:Poland,2004-2010.
(35)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09,RevisionA.01;Gaussian Inc.:Wallingford,CT,2009.
(36)Schmidt,J.R.;Corcelli,S.A.;Skinner,J.L.J.Chem.Phys. 2004,121,8887.doi:10.1063/1.1791632
(37)Papamokos,G.V.;Demetropoulos,I.N.J.Phys.Chem.A 2004,108,7291.doi:10.1021/jp049563d
(38)Kim,Y.S.;Hochstrasser,R.M.J.Phys.Chem.B 2005,109, 6884.doi:10.1021/jp0449511
(39)Pohl,G.;Perczel,A.;Vass,E.;Magyarfalvi,G.;Tarczay,G. Phys.Chem.Chem.Phys.2007,9,4698.doi:10.1039/ b705098d
(40)Cormanich,R.A.;Rittner,R.;Buhl,M.RSC Adv.2015,5, 13052.doi:10.1039/C4RA16472E
(41)Saven,J.G.;Skinner,J.L.J.Chem.Phys.1993,99,4391.
doi:10.1063/1.466092
(42)Kubo,R.Advances in Chemical Physcis;John Wiley&Sons, Inc.:New York,2007;p 101.
(43)Han,C.;Wang,J.ChemPhysChem 2012,13,1522. doi:10.1002/cphc.v13.6
Correlation between Amide-I Spectra and Structural Features of Glycine Dipeptide
CAI Kai-Cong1,2,*ZHENG Xuan1,2LIU Ya-Nan1,2LIU Shan-Hong1,2DU Fen-Fen1,2
(1College of Chemistry and Chemical Engineering,Fujian Normal University,Fuzhou 350007,P.R.China;2Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry,Xiamen 361005,Fujian Province,P.R.China)
Structural and spectroscopic features of a model dipeptide,glycine dipeptide(GLYD),were systematically investigated in the gas phase and in aqueous solution.Normal mode analysis was performed on the representative GLYD-D2O clusters selected from molecular dynamics(MD)trajectory for the vibrational parameters of amide-I mode,which is known to be sensitive to the secondary structure of proteins.On this basis, the correlation between the vibrational spectrum and the structural features of specific groups in the polypeptide was constructed.The electrostatic potential from the solvent molecules was calculated and projected onto the backbone of GLYD,and related to the amide-I frequency difference for GLYD in gas phase and solution phase. The secondary structure-dependent normal mode amide-I frequency database was also introduced for the consideration of the possible vibrational coupling that is intrinsically included in GLYD conformers.An electrostatic frequency map with secondary structural sensitivity was then built for the fast and accurate vibrational frequency prediction of the amide-I vibrational band for polypeptides in solution.
Glycine dipeptide;Amide-I band;Vibrational spectrum;Electrostatic frequency map
1 引言
要发挥正常的物理、化学以及生物学功能,蛋白质需要通过大量氢键、范德华力和疏水作用等非共价相互作用来正确折叠形成一个特定构型。组织中特定蛋白质如果发生错误折叠,将会引起空间构象变化,进而发生自组装形成难溶的纤维聚集体,引发如阿尔茨海默病、II型糖尿病、汉庭顿舞蹈症等所谓的“蛋白质构象病”1,从而给公共卫生带来巨大压力,引发重大社会问题2。要从分子水平上研究蛋白质发挥作用的机制,需要深化对蛋白质构象态即二级结构变化的结构动力学信息的认识,了解其可能的构象态布居倾向。作为一级结构和三维空间结构之间的重要桥梁,蛋白质二级结构的预测对于理解溶液相中蛋白质空间构型、折叠机制以及蛋白质所产生的功能具有重要的理论价值,而且对于揭示蛋白质结构的改变对生物体的影响乃至预防疾病的产生有着重要的指导作用3,4。
December 24,2015;Revised:February 29,2016;Published on Web:February 29,2016.
O641
10.3866/PKU.WHXB201602291
*Corresponding author.Email:ckc1117@fjnu.edu.cn;Tel:+86-591-22868161.
The project was supported by the National Natural Science Foundation of China(21103021)and Education Department of Fujian Province of China (JA13063).
国家自然科学基金(21103021)和福建省高校杰出青年科学人才培育计划(JA13063)资助项目
