铂钌团簇及电荷对甲醇活性的理论研究
2016-09-06赵俊凤孙小丽黄旭日李吉来
赵俊凤 孙小丽 黄旭日 李吉来
(吉林大学理论化学研究所,长春130023)
铂钌团簇及电荷对甲醇活性的理论研究
赵俊凤孙小丽黄旭日李吉来*
(吉林大学理论化学研究所,长春130023)
采用密度泛函理论(DFT)对钌掺杂的铂团簇阳离子([PtnRum]+,m+n=3,n≥1)活化甲醇C―H和O―H键反应进行了理论研究;探讨了电荷对[PtnRum]团簇反应活性的影响。电荷分析表明:(1)[Pt3]+团簇中正电荷在三个Pt原子上均匀分布;掺杂Ru原子后,正电荷主要分布在Ru原子上;(2)首先活化C―H键时[PtnRum]+的反应活性比[PtnRum]明显提高;首先活化O―H键时只有[Pt3]+比[Pt3]团簇的反应活性有明显提高。本研究可为金属团簇调控的C―H键和O―H键的活化提供更深入的理解。
密度泛函理论;甲醇;键活化;电荷;反应活性
1 引言
近年来,气相条件下金属团簇与小分子之间的相互作用在实验和理论上都引起了人们的广泛关注1,2。理论研究金属团簇和小分子间的相互作用对于了解它们的作用机理,预测其活性有着重要意义。铂是一种重要的催化剂材料,具有广泛的应用3-9。理论研究方面,主要集中在其团簇的稳定态几何构型、电子结构以及活化机理等方面10-14。中性体系是研究的焦点15-28;然而对于带有电团簇的研究较少29-35,尚有很多的问题亟待解决。
甲醇是重要的工业原料,也可作为液态燃料。直接甲醇燃料电池(DMFC)被认为是当今世界清洁的动力能源,其广阔的应用前景引起了人们的高度重视36-38。铂基催化剂被认为是迄今为止甲醇氧化最有效的催化剂,其中铂钌二元催化剂被认为是迄今为止最有效的DMFC阳极催化剂39-41。在铂钌二元催化剂中,钌的加入对铂催化剂性能有较好的改善,表现在:一是钌将部分d电子传递给铂,减弱铂和CO之间相互作用,有效地改善催化剂CO中毒;二是增加催化剂表面含氧物种覆盖度。在甲醇的活化反应中,C―O键活化比较困难,因此C―H和O―H键的活化通常是活化反应的初始步骤。在我们之前的工作中,深入研究了中性PtnRum(m+n=3,n≥1)团簇活化甲醇中的C―H和O―H键反应活性和机理,运用前线轨道分析了初始C―H和O―H键活化的电子转移细节27。因此,在以前研究基础上,本文中我们将深入地开展了铂钌团簇阳离子活化甲醇的理论研究。重点集中在:(1)C―H键和O―H键活化过程以及电子转移过程;(2)溶剂效应;(3)对比中性体系,探讨了电荷效应。本文对C―H键和O―H键的活化机制进行了详细的理论探索,辅以电子结构分析,丰富了对过渡金属催化C―H键和O―H键的活化的认识,对改进、设计催化剂提供可靠的理论依据。
2 计算方法
本文所有计算采用Gaussian 09程序42。采用密度泛函理论B3LYP方法43,对Ru和Pt采用双ζ价电子基组和相应的Los Alamos有效核势LANL2DZ基组44,45,对C、H和O原子选用6-31G(d)基组确定势能面上的各驻点,优化的稳定体和过渡态通过频率分析计算来证实其正确性并得到热力学信息。近期的一些方法的基准研究表明对于过渡金属体系中涉及金属氢键(M―H)、金属碳键(M―C)、金属氧键(M―O),B3LYP泛函具有很强的适用性46-49。本文所有过渡态都有唯一虚频50,通过振动模式分析进一步确认了过渡态的真实性。用内禀坐标(IRC)51计算来验证反应路径的正确连接。为了得到更为准确的反应路径的能量信息,选用B3LYP/def2-QZVPP理论水平计算单点能构建精确的势能面。溶剂化模型采用SMD模型52。为掌握电子转移细节,加深对反应的理解,对在最小能量路径上选择代表性点进行准束缚分子轨道(QRO)分析53-57。分子轨道图使用Chimera软件绘制58。
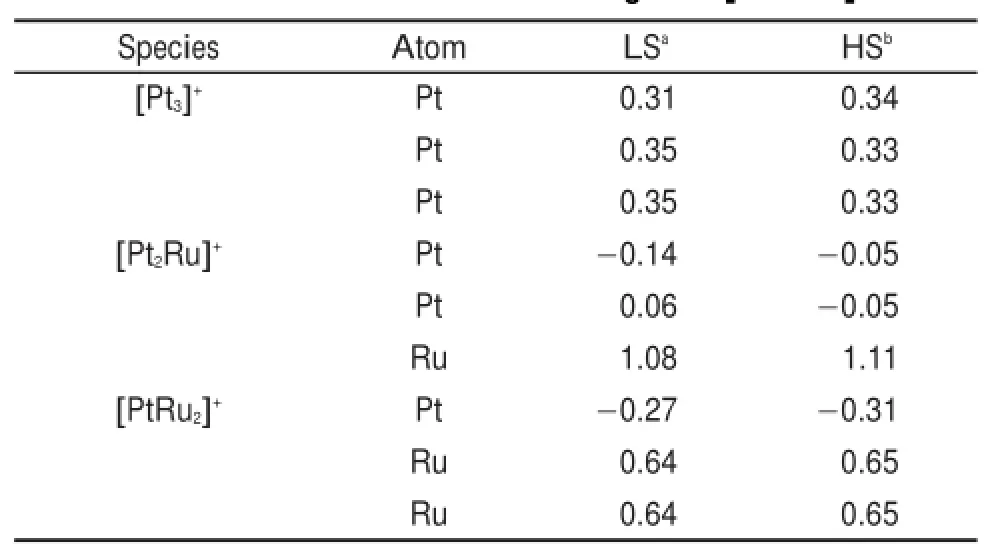
表1 [PtnRum]+的Mulliken电荷Table 1 Mulliken atomic charges of[PtnRum]+
3 结果与讨论
3.1[PtnRum]+/0团簇及其与甲醇形成的络合物
[PtnRum](m+n=3,n≥1)团簇随着Ru原子数目的增加,团簇的稳定状态趋于高自旋27,这也适用于[PtnRum]+团簇。[Pt3]+二重态是基态,[Pt2Ru]+和[PtRu2]+基态时的自旋态分别为六重态和八重态,[PtnRum]+不同自旋态下的能量和B3LYP水平下优化的几何结构见表S1和图S1(见Supporting Information)。表1是[PtnRum]+的Mulliken电荷分布,从表1中看出[Pt3]+团簇中正电荷在三个Pt原子上几乎平均分布;掺杂Ru原子后,[Pt2Ru]+团簇中正电荷主要分布在Ru原子上,电荷对Pt原子的影响不大;掺杂两个Ru原子后,[PtRu2]+团簇正电荷分布在两个Ru原子上,且使得Pt原子带一定的负电性。CH3OH和[PtnRum]+/0生成反应络合物:金属中心与甲醇的O作用形成反应复合物RC_O;金属中心与甲醇的甲基H相互作用形成反应复合物RC_C。图1表示的是CH3OH和[PtnRum]+/0生成反应络合物释放的能量。图中可以看出:(1)生成络合物RC_O释放的能量总是比生成RC_C释放的能量多;(2) CH3OH与[PtnRum]+生成的络合物比其与[PtnRum]生成的络合物稳定;(3)不论是中性[PtnRum]团簇还是阳离子[PtnRum]+,与甲醇络合释放的能量总是随着掺杂Ru原子数目的增加而减少。表S2(见Supporting Information)是[PtnRum]+/0和其与甲醇形成的反应络合物的键长参数。从中可以看出电荷对[PtnRum]+/0键长变化的影响没有规律性,但是电荷对于反应络合物中Pt―O和Pt―H键长变化的影响具有明显的规律性。除了8RC_C,[PtnRum]+和甲醇形成的反应络合物中Pt―O和Pt―H键长都比中性[PtnRum]团簇与甲醇形成的络合物中相应的键长短。这说明[PtnRum]+对甲醇的吸附能力比[PtnRum]强。以往的研究也证明正电荷体系比中性体系在反应热力学上具有优势59。

图1 [PtnRum]+/0与甲醇络合释放的能量Fig.1 Binding energies of CH3OH and[PtnRum]+/0clusters
3.2[PtnRum]+与甲醇反应
[PtnRum]+(m+n=3,n≥1)团簇活化甲醇反应两种机制如示意图1所示:M1为首先活化甲醇中的O―H键,生成中间体[M]―OCH3,再进行C―H键的活化;M2为首先活化甲醇中的C―H键,生成中间体[M]―CH2OH,进行O―H键的活化。[PtnRum]+活化O―H键时有两种机制:(a)单金属中心机制,一个Pt金属原子直接参与活化反应的反应机制,TSOa为相应过渡态,过渡态结构中有Pt…O…H三元环;(b)双金属中心机制,两个金属原子同时参与活化反应的反应机制,TSOb为相应过渡态,过渡态结构中有Pt…O…H…M(M=Pt, Ru)四元环(图2)。活化C―H时仅存在一个Pt金属原子参与C―H键的活化反应的反应机制,TSCa为相应过渡态,过渡态结构中有Pt…C…H三元环(图3)。尽管Ru也可以作为反应活性中心,我们计算了[Pt3]+掺杂一个Ru原子后,以Ru为活性中心活化C―H键和O―H的过渡态能量,从表S3(见Supporting Information)可以看出以Ru原子为活性中心的过渡态能量比以Pt原子为活性中心的能量高。在本文中我们只考虑以Pt原子为活性中心的反应。
图2为[Pt3]+(S=1/2,3/2),[Pt2Ru]+(S=3/2,5/ 2)和[PtRu2]+(S=5/2,7/2)与甲醇反应M1机制的焓变势能面。[Pt3]+活化O―H键时,双金属中心机制较为有利,2TSOb能量为-33.9 kJ∙mol-1;[Pt2Ru]+和[PtRu2]+活化O―H键时采用单个Pt原子参与活化的活化机制有利,[Pt2Ru]+活化O―H键过渡态6TSOa的能量为20.5 kJ∙mol-1,[PtRu2]+活化O― H键过渡态8TSOa的能量为65.7 kJ∙mol-1。C―H键活化过程中是单个Pt原子参与活化,过渡态为TSOaHCa。M1机制中[Pt3]+反应活性比[Pt2Ru]+和[PtRu2]+高。图3是[Pt3]+,[Pt2Ru]+和[PtRu2]+与甲醇反应M2机制的焓变势能面。M2机制中甲醇与[PtnRum]+形成反应络合物RC_C,[PtRu2]+与甲醇反应六重态时没有得到RC_C,只存在O原子与Pt原子相互作用的复合物6RC_O。C―H键活化的过渡态为TSCa,[Pt3]+活化C―H键,二重态这个过程几乎是无垒的。[Pt3]+活化O―H键存在两种反应机制:(a)单金属中心机制,通过过渡态TSCaHd将H原子转移到相邻的Pt原子上,然后再进行O―H键的活化;(b)双金属中心机制,直接进行O―H键的活化,TSCaOb为相应过渡态。图3A看出[Pt3]+活化O―H键采取单金属中心机制进行。[Pt2Ru]+和[PtRu2]+活化O―H键有两种反应机制:单金属中心机制(过渡态TSCaOa)和双金属中心机制(过渡态TSCaOb)。[Pt2Ru]+活化O―H键,四重态双金属中心机制TSCaOb有利;[PtRu2]+活化O―H键六重态单金属中心机制TSCaOa有利。在M2机制中,[Pt2Ru]+团簇与甲醇的反应活性高于[Pt3]+和[PtRu2]+团簇。图 S2-S7(见 Supporting Information)显示了[PtnRum]+团簇活化甲醇反应的M1和M2机制涉及的几何结构。对比图2和图3,[Pt3]+、[Pt2Ru]+和[PtRu2]+活化甲醇都是M2机制占优势,即反应以C―H键活化为初始步骤。前人的研究也表明甲醇在Pt表面分解反应是以C―H键的断裂为反应的初始步骤的60,61。前线轨道分析表明[PtnRum]+活化初始的O―H/C―H键的过程是质子转移(PT)的过程(见图S8-S14)。
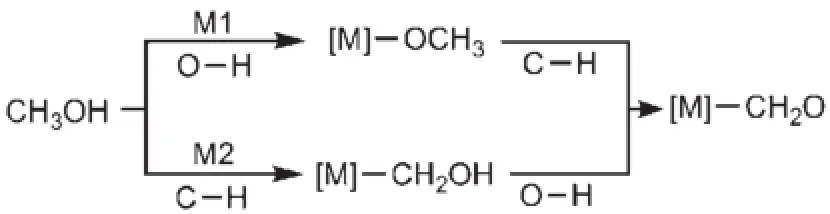
示意图1 [PtnRum]+团簇活化甲醇反应两种机制Scheme 1 Two mechanisms of reactions of CH3OH with[PtnRum]+cluster
综合[Pt3]+、[Pt2Ru]+和[PtRu2]+与甲醇反应的M1 和M2的势能面,气相下三种团簇阳离子的反应活性顺序为[Pt2Ru]+>[Pt3]+>[PtRu2]+。表S4、S5和S6(见Supporting Information)中列出了气相、介电常数ε=4、乙腈溶剂(ε=36.6)和水(ε=78.4)条件下的相对焓变。表2是[PtnRum]+(n+m=3,n≥1)与甲醇反应的M1、M2机制中优势反应路径中决速能垒62。从表中可以看出,[Pt3]+和[PtRu2]+团簇在溶剂下M1机制的能垒都比气相下的低,M2机制中则变化不大,说明溶剂对[Pt3]+和[PtRu2]+团簇与甲醇反应的以O―H键为活化初始步骤的机制有利;[Pt2Ru]+团簇与甲醇的反应在溶剂下M1和M2机制的能垒都比气相下能垒低,说明溶剂下[Pt2Ru]+对甲醇的反应活性比气相下反应活性高。表2中得到乙腈溶剂(ε=36.6)、和水(ε=78.4)中反应活性顺序为:[Pt2Ru]+>[Pt3]+>[PtRu2]+,介电常数ε=4时反应活性顺序为:[Pt2Ru]+>[PtRu2]+>[Pt3]+。

图2 B3LYP水平下气相[Pt3]+(A),[Pt2Ru]+(B)和[PtRu2]+(C)与CH3OH反应的以O―H键活化为初始步骤(M1)的ΔHg势能图Fig.2 Potential energy surface(ΔHg)for the reactions of[Pt3]+(A),[Pt2Ru]+(B),and[PtRu2]+(C)with CH3OH starting from O―H bond activation at the B3LYPlevel of theory in gas phase
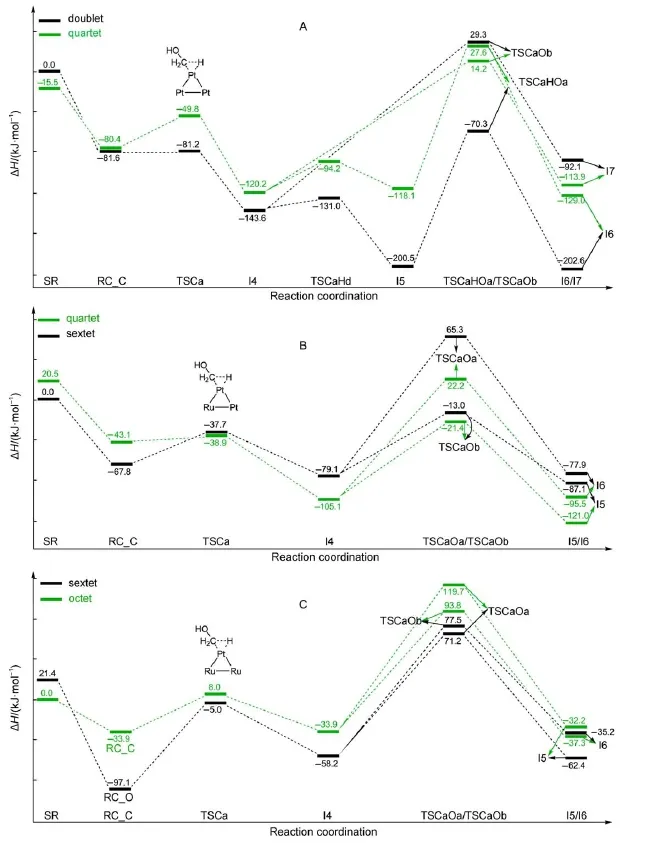
图3 B3LYP水平下气相[Pt3]+(A),[Pt2Ru]+(B)和[PtRu2]+(C)与CH3OH反应的以C―H键活化为初始步骤(M2)的ΔHg势能图Fig.3 Potential energy(ΔHg)surface for the reactions of[Pt3]+(A),[Pt2Ru]+(B)and[PtRu2]+(C)with CH3OH starting from C―H bond activation at the B3LYPlevel of theory in gas phase
3.3电荷对[PtnRum]+/0团簇活性影响
上文中提到[PtnRum]+团簇与甲醇生成反应络合物过程比其中性[PtnRum]团簇与甲醇反应释放的热量多,尤其是RC_O的形成过程中。而在C―H键和O―H键活化的活化过程中,电荷对三种团簇反应能量的影响是不相同的。表3是[PtnRum]+/0与甲醇反应首先活化O―H键和首先活化C―H键的过渡态能量。气相下:[Pt3]团簇带一个正电荷后,不论是活化O―H键还是C―H键的过渡态能量都有很大幅度的降低;[Pt2Ru]+团簇和[PtRu2]+团簇与其相应的中性团簇相比活化O―H键的过渡态能量稍有增大,但活化C―H键的过渡态能量是降低的。图4中可以明显看出[PtnRum]+团簇首先活化甲醇中的C―H键比中性[PtnRum]团簇活性高,是因为催化剂上电荷越正,越有利于C―H上的H转移17,63;因活化方式不同则活化O―H键时则不一定64,65。
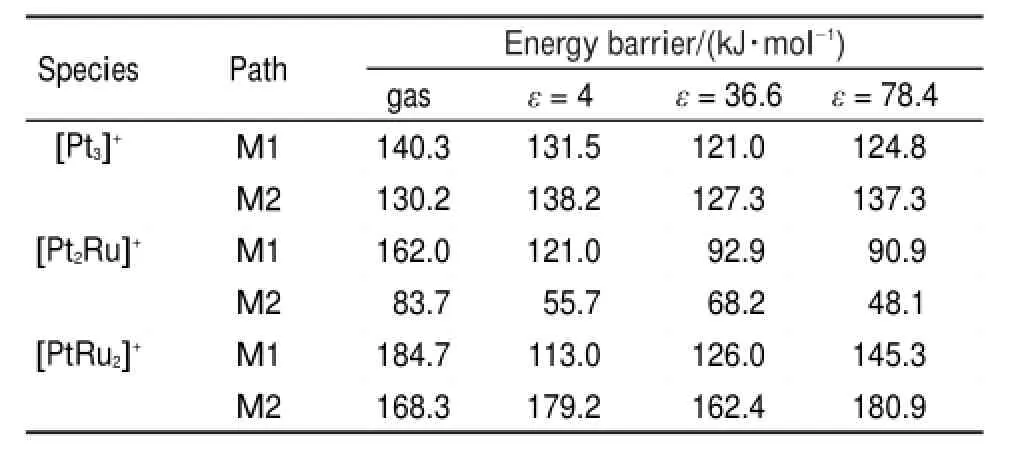
表2 B3LYP水平下标题反应中M1、M2机制的优势反应路径中决速能垒Table 2 Calculated energy barriers(kJ∙mol-1)for ratedetermining steps of the preferable pathways in M1 and M2 mechanisms for the title reaction at the B3LYPlevel

表3 气相下[PtnRum]+/0与甲醇反应活化O―H键和C―H键的过渡态能量Table 3 Transition state energies of O―H and C―H bond activation for the reactions of[PtnRum]+/0and CH3OH in gas phase
电荷的影响不仅仅反映在反应能量上,对反应机制也有影响。[PtnRum]+与中性[PtnRum]活化甲醇的反应相比:相同点是二者都是以C―H键的活化为反应的初始步骤,即M2机制有利;初始步骤中C―H和O―H键的活化都是质子转移。不同点是后者O―H键活化方式与活化的先后顺序有关。如果O―H键首先被活化,单金属中心机制有利;如果先活化C―H键,再活化O―H键,则双金属中心机制有利,而前者O―H键活化方式的选择与其被活化的先后顺序没有明显关系:在M1机制中,[Pt3]+活化O―H键采用双金属中心机制有利,而[Pt2Ru]+和[PtRu2]+采用单金属中心机制有利;M2机制中[Pt3]+和[PtRu2]+团簇活化O―H键采用单金属中心机制,[Pt2Ru]+活化O―H键双金属中心机制有利。
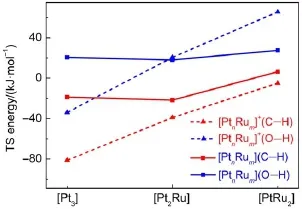
图4 气相下[PtnRum]+/0与甲醇反应首先活化O―H键和C―H键的过渡态(TS)能量Fig.4 Transition state(TS)energies of the initial O―H and C―H bond activation for the reactions of[PtnRum]+/0with CH3OH in gas phase
4 结论
在密度泛函理论水平下研究了铂钌二元团簇阳离子[PtnRum]+(n+m=3,n≥1)活化甲醇的反应,探讨了电荷、掺杂金属、溶剂等对团簇反应活性的影响。可归结出以下结论:(1)三种团簇与甲醇的反应活性顺序为[Pt2Ru]+>[Pt3]+>[PtRu2]+;(2)气相下,[PtnRum]+与甲醇的活化反应是以C―H键活化为初始步骤;(3)溶剂极性会提高[PtnRum]+对甲醇的反应活性;但极性大小对不同掺杂体系影响不同;(4)电荷对PtnRum活化C―H键和O―H键的影响存在明显不同:加入电荷后[PtnRum]+首先活化C―H键的活性明显提高,而活化O―H键时则[Pt3]+比[Pt3]团簇的反应活性有明显提高,[Pt2Ru]+和[PtRu2]+团簇首先活化O―H键的反应活性与其对应的中性团簇相比有所降低。本文的研究可为铂钌二元金属团簇活性研究提供理论依据。
Supporting Information:Enthalpies,optimized geometries and schematic FMO diagram involved in the reaction of [PtnRum]+(n+m=3,n≥1)with CH3OH have been included. This information is available free of charge via the internet at http://www.whxb.pku.edu.cn.
References
(1)Tartaglino,U.;Zykova-Timan,T.;Ercolessi,F.;Tosatti,E. Phys.Rep.2005,411,291.doi:10.1016/j.physrep.2005.01.004
(2)Knickelbein,M.B.Ann.Rev.Phys.Chem.1999,50,79.
doi:10.1146/annurev.physchem.50.1.79
(3)Wen,Z.;Liu,J.;Li,J.Adv.Mater.2008,20,743.doi:10.1002/ adma.200701578
(4)Achatz,U.;Berg,C.;Joos,S.;Fox,B.S.;Beyer,M.K.;Niedner-Schatteburg,G.;Bondybey,V.E.Chem.Phys.Lett. 2000,320,53.doi:10.1016/S0009-2614(00)00179-2
(5)Kwon,Y.H.;Kim,S.C.;Lee,S.Y.Macromolecules 2009,42, 5244.doi:10.1021/ma900781c
(6)Li,Y.;Tang,L.;Li,J.Electrochem.Commun.2009,11,846.
doi:10.1016/j.elecom.2009.02.009
(7)Jeon,M.K.;Daimon,H.;Lee,K.R.;Nakahara,A.;Woo,S.I. Electrochem.Commun.2007,9,2692.doi:10.1016/j. elecom.2007.09.001
(8)Liu,Y.C.;Qiu,X.P.;Huang,Y.Q.;Zhu,W.T.J.Power Sources 2002,111,160.doi:10.1016/S0378-7753(02)00298-7
(9)Martínez-Huerta,M.V.;Rodríguez,J.L.;Tsiouvaras,N.;Peña, M.A.;Fierro,J.L.G.;Pastor,E.Chem.Mater.2008,20,4249.
doi:10.1021/cm703047p
(10)Tian,W.Q.;Ge,M.;Sahu,B.R.;Wang,D.;Yamada,T.; Mashiko,S.J.Phys.Chem.A 2004,108,3806.doi:10.1021/ jp0498365
(11)Xiao,L.;Wang,L.J.Phys.Chem.A 2004,108,8605.
doi:10.1021/jp0485035
(12)Majumdar,D.;Dai,D.;Balasubramanian,K.J.Chem.Phys. 2000,113,7919.doi:10.1063/1.1316039
(13)Majumdar,D.;Dai,D.;Balasubramanian,K.J.Chem.Phys. 2000,113,7928.doi:10.1063/1.1316009
(14)Grönbeck,H.;Andreoni,W.Chem.Phys.2000,262,1. doi:10.1016/S0301-0104(00)00294-9
(15)de Visser,S.P.;Shaik,S.J.Am.Chem.Soc.2003,125,7413.
doi:10.1021/ja034142f
(16)Geng,C.;Ye,S.;Neese,F.Angew.Chem.Int.Edit.2010,49, 5717.doi:10.1002/anie.v49:33
(17)Li,J.;Wu,X.N.;Schlangen,M.;Zhou,S.;González-Navarrete,P.;Tang,S.;Schwarz,H.Angew.Chem.Int.Edit. 2015,54,5074.doi:10.1002/anie.v54.17
(18)Li,J.L.;Geng,C.Y.;Huang,X.R.;Zhang,X.;Sun,C.C. Organometallics 2007,26,2203.doi:10.1021/om070039d
(19)Li,J.L.;Zhang,X.;Huang,X.R.Phys.Chem.Chem.Phys. 2012,14,246.doi:10.1039/C1CP22187F
(20)Schwarz,H.Angew.Chem.Int.Edit.2011,50,10096.doi: 10.1002/anie.201006424
(21)Shaik,S.;Cohen,S.;Wang,Y.;Chen,H.;Kumar,D.;Thiel,W. Chem.Rev.2009,110,949.doi:10.1021/cr900121s
(22)Shaik,S.;de Visser,S.P.;Ogliaro,F.;Schwarz,H.;Schröder, D.Curr.Opin.Chem.Biol.2002,6,556.doi:10.1016/S1367-5931(02)00363-0
(23)Shaik,S.;Kumar,D.;de Visser,S.P.;Altun,A.;Thiel,W. Chem.Rev.2005,105,2279.doi:10.1021/cr030722j
(24)Sun,X.;Li,J.;Huang,X.;Sun,C.Curr.Inorg.Chem.2012,2, 64.doi:10.2174/1877944111202010064
(25)Ye,S.;Neese,F.Curr.Opin.Chem.Biol.2009,13,89.
doi:10.1016/j.cbpa.2009.02.007
(26)Ye,S.;Neese,F.Proc.Natl.Acad.Sci.U.S.A.2011,108, 1228.doi:10.1073/pnas.1008411108
(27)Zhao,J.F.;Sun,X.L.;Li,J.L.;Huang,X.R.Acta Phys.-Chim.Sin.2015,31,1077.[赵俊凤,孙小丽,李吉来,黄旭日.物理化学学报,2015,31,1077.]doi:10.3866/PKU. WHXB201504014
边界层采用YSU方案,陆面过程及长、短波辐射方案分别为:5层热力辐散方案、RRTM方案和Dudhia方案。36 km和12 km均采用Lin微物理方案和Kain-Fritsch积云对流参数化方案。
(28)Zhong,W.;Liu,Y.;Zhang,D.J.Mol.Model.2012,18,3051.
doi:10.1007/s00894-011-1318-7
(29)Koszinowski,K.;Schlangen,M.;Schröder,D.;Schwarz,H. Int.J.Mass Spectrom.2004,237,19.doi:10.1016/j. ijms.2004.06.009
(30)Koszinowski,K.;Schröder,D.;Schwarz,H.Chem.Phys. Chem.2003,4,1233.doi:10.1002/cphc.200300840
(31)Koszinowski,K.;Schröder,D.;Schwarz,H.J.Am.Chem.Soc. 2003,125,3676.doi:10.1021/ja029791q
(32)Koszinowski,K.;Schröder,D.;Schwarz,H.Organometallics 2003,22,3809.doi:10.1021/om030272l
(33)Koszinowski,K.;Schröder,D.;Schwarz,H.J.Phys.Chem.A 2003,107,4999.doi:10.1021/jp027713j
(34)Koszinowski,K.;Schröder,D.;Schwarz,H.Angew.Chem.Int. Edit.2004,43,121.doi:10.1002/anie.200352817
(35)Koszinowski,K.;Schröder,D.;Schwarz,H.Organometallics 2004,23,1132.doi:10.1021/om0306675
(36)Kamarudin,S.K.;Achmad,F.;Daud,W.R.W.Int.J.Hydrog. Energy 2009,34,6902.doi:10.1016/j.ijhydene.2009.06.013
(37)Kamarudin,S.K.;Daud,W.R.W.;Ho,S.L.;Hasran,U.A. J.Power Sources 2007,163,743.doi:10.1016/j. jpowsour.2006.09.081
(38)Rabis,A.;Rodriguez,P.;Schmidt,T.J.ACS Catal.2012,2, 864.doi:10.1021/cs3000864
(39)Jin,X.;He,B.;Miao,J.;yuan,J.;Zhang,Q.;Niu,L.Carbon 2012,50,3083.doi:10.1016/j.carbon.2012.03.004
(40)La-Torre-Riveros,L.;Guzman-Blas,R.;Méndez-Torres,A.E.; Prelas,M.;Tryk,D.A.;Cabrera,C.R.ACS Appl.Mater. Interfaces 2012,4,1134.doi:10.1021/am2018628
(41)Nishanth,K.G.;Sridhar,P.;Pitchumani,S.;Shukla,A.K.Fuel Cells 2012,12,146.doi:10.1002/fuce.201100113
(42)Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09,RevisionA.02;Gaussian Inc.:Wallingford,CT,2009.
(43)Becke,A.D.J.Chem.Phys.1993,98,5648.doi:10.1063/ 1.464913
(44)Hay,P.J.;Wadt,W.R.J.Chem.Phys.1985,82,270.
doi:10.1063/1.448799
(45)Hay,P.J.;Wadt,W.R.J.Chem.Phys.1985,82,299.
doi:10.1063/1.448975
(46)Li,J.;Ryde,U.Inorg.Chem.2014,53,11913.doi:10.1021/ ic5010837
(47)Li,J.L.;Mata,R.A.;Ryde,U.J.Chem.Theory Comput. 2013,9,1799.doi:10.1021/ct301094r
(48)Zhang,X.;Schwarz,H.Chem.Eur.J.2010,16,5882. doi:10.1002/chem.201000567
(49)Zhang,X.;Schwarz,H.Theor.Chem.Acc.2011,129,389. doi:10.1007/s00214-010-0861-0
(50)Weigend,F.;Ahlrichs,R.Phys.Chem.Chem.Phys.2005,7, 3297.doi:10.1039/b508541a
(51)Fukui,K.J.Phys.Chem.1970,74,4161.doi:10.1021/ j100717a029
(52)Marenich,A.V.;Cramer,C.J.;Truhlar,D.G.J.Phys.Chem.B 2009,113,6378.doi:10.1021/jp810292n
(53)Neese,F.J.Am.Chem.Soc.2006,128,10213.doi:10.1021/ ja061798a
(54)Neese,F.WIREs Comput.Mol.Sci.2012,2,73.doi:10.1002/ wcms.81
(55)Sun,X.;Geng,C.;Huo,R.;Ryde,U.;Bu,Y.;Li,J.J.Phys. Chem.B 2014,118,1493.doi:10.1021/jp410727r
(56)Sun,X.H.;Sun,X.L.;Geng,C.Y.;Zhao,H.T.;Li,J.L. J.Phys.Chem.A 2014,118,7146.doi:10.1021/jp505662x
(57)Sun,X.L.;Huang,X.R.;Li,J.L.;Huo,R.P.;Sun,C.C. J.Phys.Chem.A 2012,116,1475.doi:10.1021/jp2120302
(58)Pettersen,E.F.;Goddard,T.D.;Huang,C.C.;Couch,G.S.; Greenblatt,D.M.;Meng,E.C.;Ferrin,T.E.J.Comput. Chem.2004,25,1605.doi:10.1002/jcc.20084
(59)Li,J.;González-Navarrete,P.;Schlangen,M.;Schwarz,H. Chem.Eur.J.2015,21,7780.doi:10.1002/chem.v21.21
(60)Greeley,J.;Mavrikakis,M.J.Am.Chem.Soc.2002,124, 7193.doi:10.1021/ja017818k
(61)Greeley,J.;Mavrikakis,M.J.Am.Chem.Soc.2004,126, 3910.doi:10.1021/ja037700z
(62)Kozuch,S.;Shaik,S.Accounts Chem.Res.2010,44,101. doi:10.1021/ar1000956
(63)Li,J.;Wu,X.N.;Zhou,S.;Tang,S.;Schlangen,M.;Schwarz, H.Angew.Chem.Int.Edit.2015,54,12298.doi:10.1002/ anie.201503763
(64)Li,J.;Zhou,S.;Wu,X.N.;Tang,S.;Schlangen,M.;Schwarz, H.Angew.Chem.Int.Edit.2015,54,11861.doi:10.1002/ anie.201505336
(65)Zhong,W.H.;Zhang,D.J.Prog.React.Kinet.Mech.2013, 38,86.doi:10.3184/146867813X13590434110230
A Theoretical Study on the Reactivity and Charge Effect of PtRu Clusters toward Methanol Activation
ZHAO Jun-FengSUN Xiao-LiHUANG Xu-RiLI Ji-Lai*
(Institute of Theoretical Chemistry,Jilin University,Changchun 130023,P.R.China)
Density functional theory(DFT)calculations were performed to gain mechanistic insight into the methanol C―H and O―H bond activations mediated by ruthenium-doped platinum cationic clusters[PtnRum]+(m+n=3,n≥1).The charge effect on the reactivity has been elucidated.Calculations show that positive charge is evenly distributed on the three Pt atoms of the[Pt3]+cluster,while in the Ru-doped clusters,positive charge is mainly distributed on the Ru atom(s).The reactivity of[PtnRum]+is significantly greater than neutral [PtnRum]during the initial C―H bond cleavage,while only[Pt3]+exhibits greater reactivity than[Pt3]in the course of O―H bond cleavage.This study may aid in deeper understanding of C―H/O―H bond activations mediated by metal clusters.
Density functional theory;Methanol;Bond activation;Charge;Reactivity
October 6,2015;Revised:February 22,2016;Published:Published on Web:February 22,2016.
O641
10.3866/PKU.WHXB201602221
*Corresponding author.Email:jilai@jlu.edu.cn.
The project was supported by the National Key Basic Research Program of China(973)(2012CB932800)and National Natural Science Foundation of China(21103064,21473070).
国家重点基础研究发展规划项目(973)(2012CB932800)和国家自然科学基金(21103064,21473070)资助
