头孢噻肟钠制备工艺的改进
2016-09-05孙津鸽高得瀛刘红坤李文杰李志军
孙津鸽,高得瀛,刘红坤,李文杰,李志军
(河南康达制药有限公司,河南 周口 466200)
头孢噻肟钠制备工艺的改进
孙津鸽,高得瀛,刘红坤,李文杰,李志军
(河南康达制药有限公司,河南 周口 466200)
为改进头孢噻肟钠制备工艺。在混合溶媒中,头孢噻肟酸与成盐剂反应制得头孢噻肟钠溶液,再滴加溶析剂析出头孢噻肟钠固体。制得头孢噻肟钠的含量稳定在96%以上,且目标产物经质谱和核磁共振确认结构。改进后的工艺优化了反应条件,提高了含量,降低了成本,更适合工业化生产。
头孢噻肟酸;头孢噻肟钠;成盐剂;工艺改进
头孢噻肟钠(Cefotaxime sodium)又名头孢菌素钠,为白色、类白色或淡黄白色结晶,无臭或微有特殊臭味。其化学名为(6R,7R)-3-[(乙酰氧基)甲基]-7-[(2-氨基-4-噻唑基)-(甲氧亚氨基)乙酰氨基]-8-氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯- 2-甲酸钠盐,为第三代头孢类半合成广谱抗生素,是由德国赫司特公司、法国鲁塞尔公司、日本中外制药开发出的一个头孢类产品[1]。该药具有抗菌谱广,抗菌作用强,毒副作用小,对β- 内酰胺酶稳定等特点,对溶血性链球菌、肺炎球菌、流感杆菌及脑膜炎球菌等有极高的抗菌活力,尤其是对肠杆菌作用强,并对大多数厌氧菌有强抑制作用;临床广泛应用于对治疗敏感细菌引起的败血症、化脓性脑膜炎及呼吸道、泌尿道、胆道、骨和关节、皮肤和软组织、腹腔、消化道、五官、生殖器等部位的感染,也可用于免疫功能低下、抗体细胞减少等防御功能低下的感染性疾病[2-3]。
头孢噻肟钠的制备工艺一般分两步操作:第一步是以7-ACA与AE-活性酯为原料,在惰性有机溶剂(如二氯甲烷等溶剂)中,加入三乙胺及相转移催化剂合成头孢噻肟酸;第二步头孢噻肟酸与成盐剂反应得头孢噻肟钠,然后加溶析剂结晶制得头孢噻肟钠成品,所用成盐剂主要有三水合乙酸钠、碳酸氢钠、甲酸钠等,结晶溶剂主要有乙醇、丙酮、异丙醇等[3-6]。
就目前的工艺现状而言,在头孢噻肟钠结晶过程中极易出现聚结成胶现象,结晶过程极不稳定,并且还存在头孢噻肟钠有效含量偏低等问题。这些问题的存在一方面导致产品质量较差且不稳定,另外还在一定程度上导致生产成本的提高及原材料的浪费,最重要的是在药物生产过程中,常常由于药物含量偏低,微量毒副作用物质的存在使药物的应用达不到应有效果[3]。
鉴于国内头孢噻肟钠制备的生产现状,改进头孢噻肟钠制备工艺,提高产品质量与品级,并有效的降低生产成本与能耗,不仅具有十分重要的现实意义和应用价值,而且成为提高我国头孢菌素类抗生素在国际上的竞争力的重中之重。针对我国制药工业头孢噻肟钠生产中存在的产品纯度低,分离困难且结晶过程中易聚结成胶等问题,本课题深入系统的研究了头孢噻肟钠成盐反应和结晶过程,改进头孢噻肟钠制备工艺。应用该工艺生产出的产品不仅质量更好,而且收率也得到了较大提高。
1 合成路线
本研究根据参考文献[6-8]合成阿德福韦酯,并进行了工艺改进。本合成路线如下:
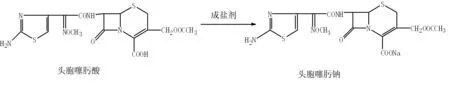
2 实验部分
2.1 仪器与试剂
SPD-10AT高效液相色谱仪(日本岛津公司);DLSB-5/40 ℃低温冷却循环泵(郑州英峪予华仪器有限公司);JJ-1定时电动搅拌(江苏金坛市医疗仪器厂);FE20实验室PH计(瑞士METTLER TOLEDO);KF-1型水分测定仪(上海市安亭电子仪器厂);RY-1型熔点测定仪(天津市分析仪器厂);ACF-300型1HNMR核磁共振仪(Bruker公司);VGZAB-HS型MS质谱仪。
所用原料及试剂:头孢噻肟酸(自制,含量99.5%),所用试剂均为国内市售AR或CP级试剂。
2.2 合成步骤
本文在已有文献[3-9]基础上,进行优化实验,合成步骤如下:
250mL反应瓶中,先投注射用水10g,亚硫酸氢钠0.2g,维持温度5~10℃,搅拌溶解后,加投料丙酮50g,头孢噻肟酸20g,边搅拌边缓慢加成盐剂调节pH值至5.5~6.5,待pH值稳定后加活性炭0.2g脱色20min,过滤,用15g丙酮与5g注射用水混合液洗涤炭饼。滤液转入500mL反应瓶中,升温至20±2℃,加入二氯甲烷5g,搅拌10min,缓慢滴加丙酮约50g至料液浑浊,加入晶种0.1g,养晶30min。继续滴加结晶丙酮160g,滴加结束后,降温至5~10℃养晶1h后,过滤,用20g异丙醇洗涤滤饼,甩干。60℃真空干燥至水分<2.5%,得白色结晶性粉末,即为产品头孢噻肟钠。
2.3 试验数据
2.3.1 主要生产参数

表1 主要生产参数
2.3.2 产品主要参数
产量:19.1g, 含量大于96%,质量收率95%,m.p.:162.0-163.2℃,与文献[1]报道数据一致。1HNMRδ:2.01(s,3H),3.25~3.21(d,J=17.2HZ,1H),3.50~3.46(d,J=17.2HZ,1H),3.84(s,3H),4.79~4.77(d,J=12.0HZ,1H),5.01~4.98(d,J=12.0HZ,1H),5.03~5.02(d,J=4.8HZ,1H),5.62~5.58(d,J=8.0HZ,1H),6.73(s,1H),7.30(s,2H),9.57~9.55(d,J=8.0HZ,1H)。MS:m/z:454.05 [M-Na]-,与头孢噻肟钠构型及文献[3]报道数据一致。
3 结果讨论
头孢噻肟钠稳定性较差,其酰胺侧链、伊内酰胺环以及乙酰基三个部位都可能发生降解,在有水分子存在的条件下易被水解,碱、酸和温度升高均能促进水解,本文选择成盐料液pH值为5.5~6.5,研究[3]表明,在此pH值下,头孢噻肟钠水溶液比较稳定,且后期结晶效果较好。
本文在头孢噻肟钠成盐过程中,选用五种成盐剂,试验结果表明使用混合成盐剂乙酸钠与异辛酸钠(摩尔比1:2)制得头孢噻肟钠含量最高,且收率高,虽然其他适中成盐剂制得头孢噻肟钠也符合2015版药典标准,但其质量均不如用使用混合成盐剂乙酸钠与异辛酸钠(摩尔比1:2)制得头孢噻肟钠。
本文在头孢噻肟钠结晶过程中,先加入适量二氯甲烷,搅拌形成乳化溶,再滴加丙酮,优化了头孢噻肟钠的结晶工艺,得到的头孢噻肟钠产品含量高、晶体颗粒大、粒径分布均匀,易过滤、洗涤和干燥。
改进后的工艺所用各步原料和试剂易得,优化了反应条件,提高了含量,降低了成本,具有较大的工业价值和潜在社会经济效益。
[1] 国家药典委员会.中华人民共和国药典2015年版(二部)[M].北京:化学工业出版社,2015.
[2] 赵洪娥,刘 华,蔡秋琴,等.头孢噻肟钠结晶工艺的研究[J].河北化工,2013,36(2):9-10.
[3] 张海涛.头孢噻肟钠结晶技术研究[D] .天津:天津大学,2008.
[4] 郭军臣,于秀芹.溶剂粘度对头孢噻肟钠结晶晶形的影响[J].内蒙古石油化工,2014(12):30-32.
[5] 王 彪.头孢噻肟钠成盐方法的研究[J].黑龙江科技信息,2012(6):24-24.
[6] 胡文滨.头孢噻肟钠的精制[J].河北化工,2011,34(7):6-8.
[7] 代 羿,白 波,王卫青.头孢噻肟钠缩合工艺探讨[K].辽宁医药,2008(1):17-18.
[8] 卢娥辉,郑熙展.头孢噻肟成钠盐的工艺研究[J].中国现代应用药学,2006,32(2):117-120.
[9] M.Zajac,w.Musial,L.Pawiowski.Stability of cefotaxime sodium in solid state,Pharmazie,2000,55:917-918.
(本文文献格式:孙津鸽,高得瀛,刘红坤,等.头孢噻肟钠制备工艺的改进[J].山东化工,2016,45(04):12-13.)
Improvement on Synthesis Process of Cefotaxime Sodium
Sun Jinge, Gao Deying,Liu Hongkun,Li Wenjie, Li Zhijun
(Henan Kang Da Pharmaceutical Co., Ltd.,Zhoukou 466200,China)
In order to improve the synthesis process of Cefotaxime sodium. Make solution of Cefotaxime Sodium by salifying reaction of Cefotaxime acid in the mixed solvent, and then dropping dilution agent to make Cefotaxime Sodium precipitated.The content of the prepared Cefotaxime sodium is more than 96%. The structure of adefovir dipivoxil was confirmed by MS and 1H-NMR. The research optimized the hasreaction conditions and enhanced receiving content and reduced the cost . So it more suitables for industrial production requirements.
cefotaxime acid ; cefotaxime sodium ; salt forming agent ;process optimization
2016-01-12
孙津鸽(1987—),女,河南洛阳人,硕士研究生,主要从事药物合成工艺研究。
R914.5
A
1008-021X(2016)04-0012-02
