Mg团簇表面自扩散行为的原子模拟研究
2016-09-01戴雄英杨剑瑜易国军刘艳辉
戴雄英,杨剑瑜,易国军,刘艳辉
(湖南工程学院 理学院,湘潭 411104)
Mg团簇表面自扩散行为的原子模拟研究
戴雄英,杨剑瑜,易国军,刘艳辉
(湖南工程学院 理学院,湘潭 411104)
通过研究团簇表面原子的扩散过程可以实现人工设计团簇的结构.结合分子动力学和分析型嵌入原子方法研究了Mg团簇表面自扩散行为.吸附原子跨过台阶扩散的两种机制以及Mg团簇上的5条面间自扩散路径.结果表明吸附原子在不同面上及面间扩散的扩散能力差别较大.
分子动力学;表面自扩散;团簇
0 引 言
近年来,表面扩散行为引起了广泛的关注.在理论方面,主要研究了吸附原子在表面的扩散机制,如跃迁机制[1]、交换机制[2]以及低温下的量子扩散机制[3]等.在实验中,用扫描隧道显微镜(STM)观测大团簇在基底表面的二维扩散行为[4,5].但是,这些理论和实验研究主要集中在吸附原子、空位等在完整表面或台阶的扩散,涉及到原子在团簇表面的扩散很有限.尤其对基底为HCP结构的团簇表面的扩散行为的研究不多.
我们选择HCP结构的金属镁(Mg)作为研究对象.主要源于以下两个原因,其一是镁在自然界中分布非常广泛,主要蕴藏在菱镁矿、光卤石和海水等中[6].Mg的密度很小,而比强度较高,并能与铝、铜等金属形成性能良好的合金.世界各国都在加大力度研制和开发镁及镁合金.其二是Mg的c/a接近理想值,我们课题组利用自己提出的多体势函数计算了Mg的表面能和堆垛层错能等[7],计算结果均与相关实验数据吻合得比较好.
1 理论模型与计算方法

图1 587个Mg原子组成的六角多面体结构团簇

参数AlMgAl-Mgn0.380.55F02.4630.759fe(eV/Å)2.6861.602α(10-2)0.3924.135β(10-2)-1.463kc0.7500.100ra(Å)3.185rb(Å)2.9581k0-113.8k1-18.5144.6k2-11.3-76.0k39.4632.532k4-12.814.2k561.8-5.650k6-29.40.694k-133.2rc(Å)3.017μ1.13
采用淬火的分子动力学和肘弹性波(NEB)方法计算相应过程的扩散激活能(Eb).NEB方法是估算扩散激活能和找到最小能量扩散路径的有效方法之一.吸附原子沿扩散路径移动过程中,必然有一个能量极小点(吸附点)和能量极大点(鞍点),两者之间的能量差即为吸附原子相应的扩散激活能.Eb=Esad-Emin,式中,Esad和Emin分别指吸附原子在鞍点和起始吸附点的系统总能量.
2 结果与讨论
在团簇的表面引入一个吸附原子来研究其表面的自扩散行为.分析该吸附原子的轨迹可以揭示扩散的机制.
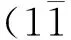

图2 Mg原子从(0001)面自扩散到相邻面的路径

图3 Mg原子01)面间自扩散的三条路径

2.1交换机制的研究
目前,金属原子的扩散机制主要分为两类,一类是跃迁机制,即吸附原子从一个平衡位置跨过势垒进入另一个新的平衡位置的扩散[8];另一类则是交换机制,即吸附原子通过替代表面原子而进行扩散.吸附原子在团簇表面的自扩散主要包括相同面内的自扩散以及从一个表面跨过台阶扩散到另一个表面的面间自扩散.吸附原子跨过台阶扩散过程中,由于近邻配位数的减少,需要克服一个额外的能量势垒,即所谓的 Ehrlich.Schwoebel(ES)势垒[9].表2为5条路径不同扩散机制所对应的ES势垒.从表中可以看出,不同的扩散路径和扩散机制对应ES势垒相差甚大.
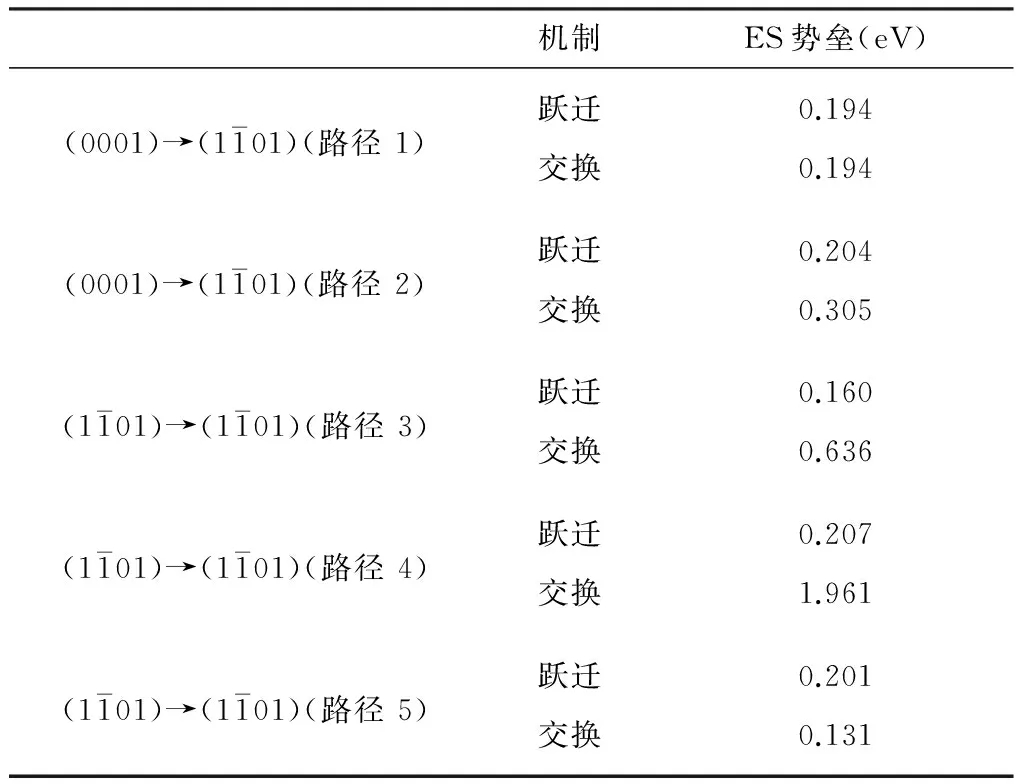
表2 采用不同扩散机制跨过台阶对应的ES势垒
从表2可以看出,吸附原子只有通过路径1和路径5实现相邻面间扩散时,与其他路径不同,路径1中,吸附原子采用跃迁和交换这两种机制所需克服的ES势垒相同,皆为0.194 eV,即两种机制基本是平权的.对路径5而言,采用跃迁机制比交换机制所需克服的ES势垒相差不大,其值分别为0.201 eV和0.131 eV.而通过其他路径扩散时,采用跃迁机制和交换机制所需克服的ES势垒相差较大,前者远小于后者.迄今为止,大部分文献报道吸附原子面间扩散时大多采用交换机制.然而,对本文研究的Mg吸附原子在其团簇表面上的面间自扩散过程中,跃迁机制却是占主导地位.这意味着Mg吸附原子很难通过与台阶上的Mg原子交换位置进入到团簇的内部.
为了详细了解吸附原子在团簇表面的自扩散行为,我们计算了每条路径对应的能量势垒曲线,如图4所示.我们对图中的横坐标进行了归一化处理,其中,0.0和1.0分别对应于吸附原子扩散过程中的始末位置.图中最小的系统能量被定义为0.
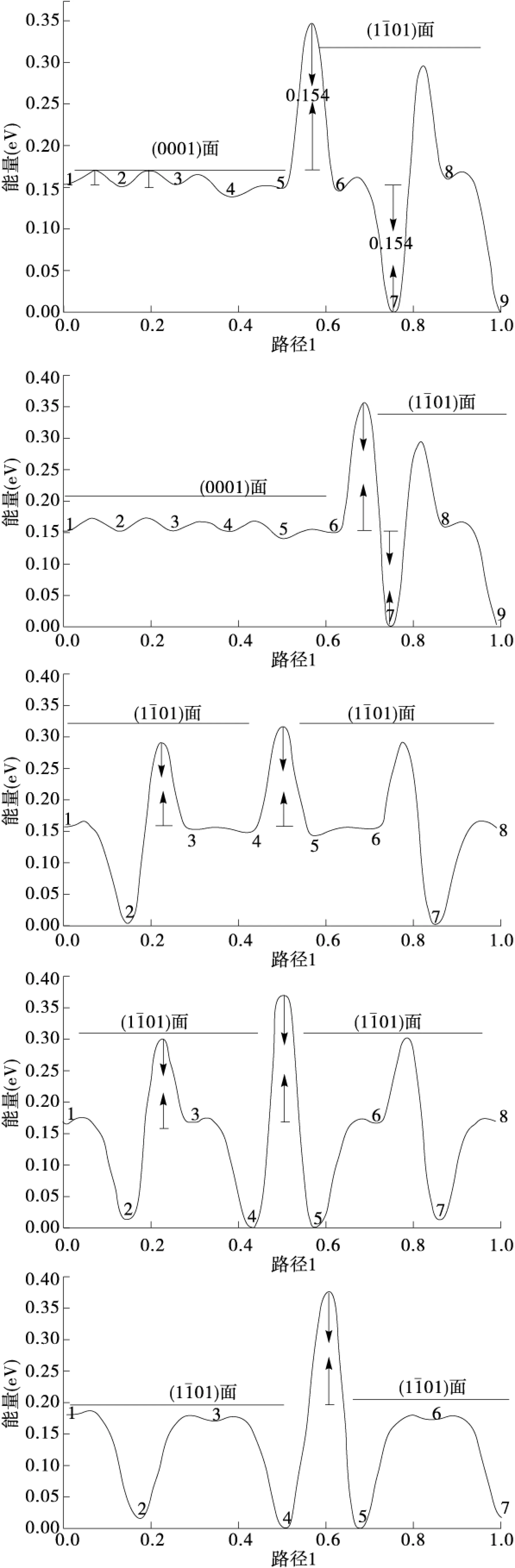
图4 Mg原子表面自扩散5条路径所对应的能量势垒曲线
3 结 论
[1] G.L. Kellogg,Field Ion Microscope Studies of Single—atom Surface Diffusion and Cluster Nucleation on Metal Surfaces[J]. Surf. Sci. Rep.,1994, 21(1-2):1-88.
[2] T.T.Tsong and C.L. Chen. Atomic Replacement and Vacancy Formation and Annihilation on Iridium Surfaces[J]. Nature, 1992, 355(23): 328-331.
[3] C. Zheng, C. Yeung, M. Loy, et al. Quantum Diffusion of H on Pt (111): Step Effects[J]. Phys. Rev. Lett., 2006, 97(16): 166101-166104.
[4] J.M. Wen, S.L. Chang, J.W. Burnett, et al. Diffusion of Large Two-Dimensional Ag Clusters on Ag (100) [J]. Phys. Rev. Lett.,1994,73(19):2591-2594.[5] L. Bardotti, P. Jenson, A. Hoareau, et al. Experimental Observation of Fast Diffusion of Large Antimony Clusters on Graphite Surfaces[J]. Phys. Rev. Lett.,1995,74(23):4694-4697.
[6] 张沣,章宗和.镁合金及其应用[M].北京:化学工业出版社,2004:1-31.
[7] W. Hu, B. Zhang, B. Huang, et al. Analytic Modified Embedded Atom Potentials for HCP Metals[J]. J. Phys.: Condens. Matter, 2001,13(6):1193-1213.
[8] G. Ayrault, G. Ehrlich. Surface Self-diffusion on an FCC Crystal: an Atomic View[J]. J. Chem. Phys., 1974,60(1):281-294.
[9] G. Ehrlich, F.G. Hudda. Atomic View of Surface Self-Diffusion: Tungsten on Tungsten[J]. J. Chem. Phys., 1966,44(3): 1039-1049.
[10] C.G. Johansen, H. Huang and T.M. Lu. Diffusion and Formation Energies of Adatoms and Vacancies on Magnesium Surfaces [J].Comp. Mater. Sci.,2009, 47(1): 121-127.
[11] Y. Han, G. Liu, B. Lee. Flat-surface, Step-edge, Facet-facet, and Facet-step Diffusion Barriers in Growth of a Pb Mesa[J]. Surf. Sci.,2008, 602(13):2284-2294.
[12] J. Yang, W. Hu, Sh. Chen. Surface Self-diffusion of Adatom on Pt Cluster with Truncated Octahedron Structure[J]. Thin Solid Films, 2010, 518(14): 4041-4045.
Atomistic Simulation of Surface Self-diffusion on Mg Clusters
DAI Xiong-ying,YANG Jian-yu,YI Guo-jun,LIU Yan-hui
(College of Science, Hunan Institute of Engineering, Xiangtan 411104, China)
Based on the study of the surface diffusion process, the structure of cluster can be artificially controlled. Surface self-diffusion behaviors of Mg cluster have been studied by using the molecular dynamics simulations and an analytic embedded-atom method. The two diffusion mechanisms for adatom acrosses the step edge and five paths in which the adatom acrosses the step edge between two neighboring facets have been discussed. We find that the adatom’s mobility is different in these diffusion procedures.
molecular dynamics; surface self-diffusion; cluster
2016-02-25
湖南省教育厅一般科研资助项目(15C0324).
戴雄英(1977- ),女,博士,研究生,讲师,研究方向:材料科学与工程.
O562.1
A
1671-119X(2016)03-0060-04
