基于密度泛函理论研究水在磷酸锂(100)表面的吸附*
2016-09-01马卫华
吕 岩,王 涛,马卫华
(南京理工大学化工学院,江苏 南京 210094)
特稿
基于密度泛函理论研究水在磷酸锂(100)表面的吸附*
吕岩,王涛,马卫华
(南京理工大学化工学院,江苏南京210094)
磷酸锂是环氧丙烷异构化反应的一种高效的催化剂,经实验发现在吸附水之后磷酸锂催化性能有所提高。为了进一步了解水吸附的影响,我们运用广义梯度近似方法(GGA-PBE)研究了水分子在Li3PO4(100)表面不同吸附位点的吸附行为。通过比较不同吸附位的吸附能和几何构型参数发现:水分子倾向于吸附在Li-Li桥位上以氧端与表面邻近的两个Li原子相互作用,而H原子与磷酸根中的O原子结合。电荷布居分析结果为水分子的电荷数减少,而Li3PO4表面的Li原子和O原子电荷数增加,表明水分子从Li3PO4表面得到电子。
密度泛函理论;水分子;磷酸锂;吸附
磷酸锂(Li3PO4)晶体是一种斜方晶系的白色结晶,在彩色荧光粉、含氮磷酸锂薄膜、光学传感器、特种玻璃以及锂离子电池正极材料具有广阔的应用前景[1-5]。其中Li3PO4作为催化剂主要应用在环氧丙烷异构化反应中,具有较高的选择性和转化率。关于Li3PO4的催化性能的研究有很多,也取得了一些进展。同作为一种环氧丙烷异构化反应研究的催化剂,Li3PO4要比纳米金催化剂更廉价,且制备烯丙醇过程中性能最好,因此在理论上研究Li3PO4催化活性的起源是有必要的。在理论研究方面,N A W Holzwarth等[6]计算了一系列的Li3PO4和Li3PS4的结构,对这些结构的稳定性能进行了分析。Y. A. Du等[7-8]详细地研究了基于Li3PO4和Li3PS4的含氮磷酸锂薄膜上的Li+的迁移,都取得了不错的进展。然而,这些理论研究研究仅仅是从结构上进行分析,并没有涉及到两相之间的变化。
水分子在固体表面上的吸附一直以来是实验和理论研究者所关注的焦点之一,在多相催化、环境保护、腐蚀和电化学等方面有着重要的应用[9-11]。许多研究报道[12-13],在吸附水之后,一些催化剂的催化性能会更好。在过去的几十年中,对水吸附在一些无机盐表面已经做了大量的理论研究[14-16]。但是,关于水分子吸附在磷酸锂表面的理论研究还未曾有报道。因此有必要对水分子在磷酸锂表面的吸附进行研究。
本文通过采用周期性密度泛函理论,系统地研究了水分子在Li3PO4(100)表面上的吸附结构和吸附能量,并用布居数和态密度分析了吸附机理。
1 计算方法
本文所有的计算均是由是基于密度泛函理论的Materials Studio软件中的CASTEP模块完成的[17],交换相关泛函采用广义梯度近似(GGA-PBE)来描述[18]。平面波的截断能量能设为500 eV。计算选用的波函数为基于Vanderbil型的超软赝势USPP[19-20]。计算模型选用具有周期性边界的平板模型(slab)。Brillouin-zone积分选用的Monkhorst-Pack方案的网格参数设置为4×3×1。模型的结构优化采用BFGS算法,在优化过程中,水分子和上四层原子进行弛豫,其余原子坐标被固定住。计算选用的真空层厚度为15 Å。优化的结果以能量、位移和力的收敛为判据,具体为:自洽场(SCF)的收敛标准1.0×10-6eV/atom,能量的容差为1.0×10-5eV/atom,原子最大位移1×10-3Å,最大步长0.3 Å。
吸附能可以由下面的公式得到:
Eads=E[(Li3PO4(100)+H2O)relaxed]-E[(Li3PO4(100))relaxed]-E[(H2O)gas]
其中E[(Li3PO4(100)+H2O)relaxed]为吸附体系的总能量,E[(Li3PO4(100))relaxed]为Li3PO4表面基底的能量,E[(H2O)gas]为气相水分子弛豫后的能量。因此,负的吸附能表示放热过程且在吸附水之后体系更加稳定。反之,正吸附能为吸热过程。
2 结果与讨论
2.1Li3PO4和气相水相关参数的计算
首先用密度泛函理论计算了Li3PO4晶胞和气相水分子的几何结构。计算得到的Li3PO4晶胞参数是a=6.288 Å, b=5.364 Å和 c=4.631 Å, α=β=γ=90°,跟实验值a=6.115 Å, b=5.239 Å和c=4.855 Å, α=β=γ=90°[21]相一致。在同样的精度下把水分子放入一个10×10×10 Å3的超胞进行优化。优化的结果为 H-O-H 键角104.5°,O-H 键长0.977 Å。跟实验测量值H-O-H键角104.5°,O-H 键长0.96~0.98 Å[22]相一致。
2.2水分子在Li3PO4(100)表面的吸附
为了得到水分子在Li3PO4(100)表面上吸附的最稳定的构型,优化之前,将水分子放置在磷酸锂表面的上方,图1给出了Li3PO4(100)表面上所有可能吸附位置。选择水分子的初始取向以平行、垂直或者倾斜放置在Li3PO4(100)表面进行结构优化,最终得到了五种稳定的吸附构型,结果见图2。五种稳定构型吸附能和结构参数列于表1。
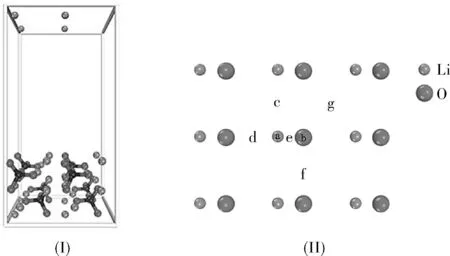
图1 Li3PO4(100)表面的侧视图(I)和俯视图(II)(只给了表层原子)Fig.1 Side view(I)and top view (Ⅱ) of the Li3PO4(100) surface
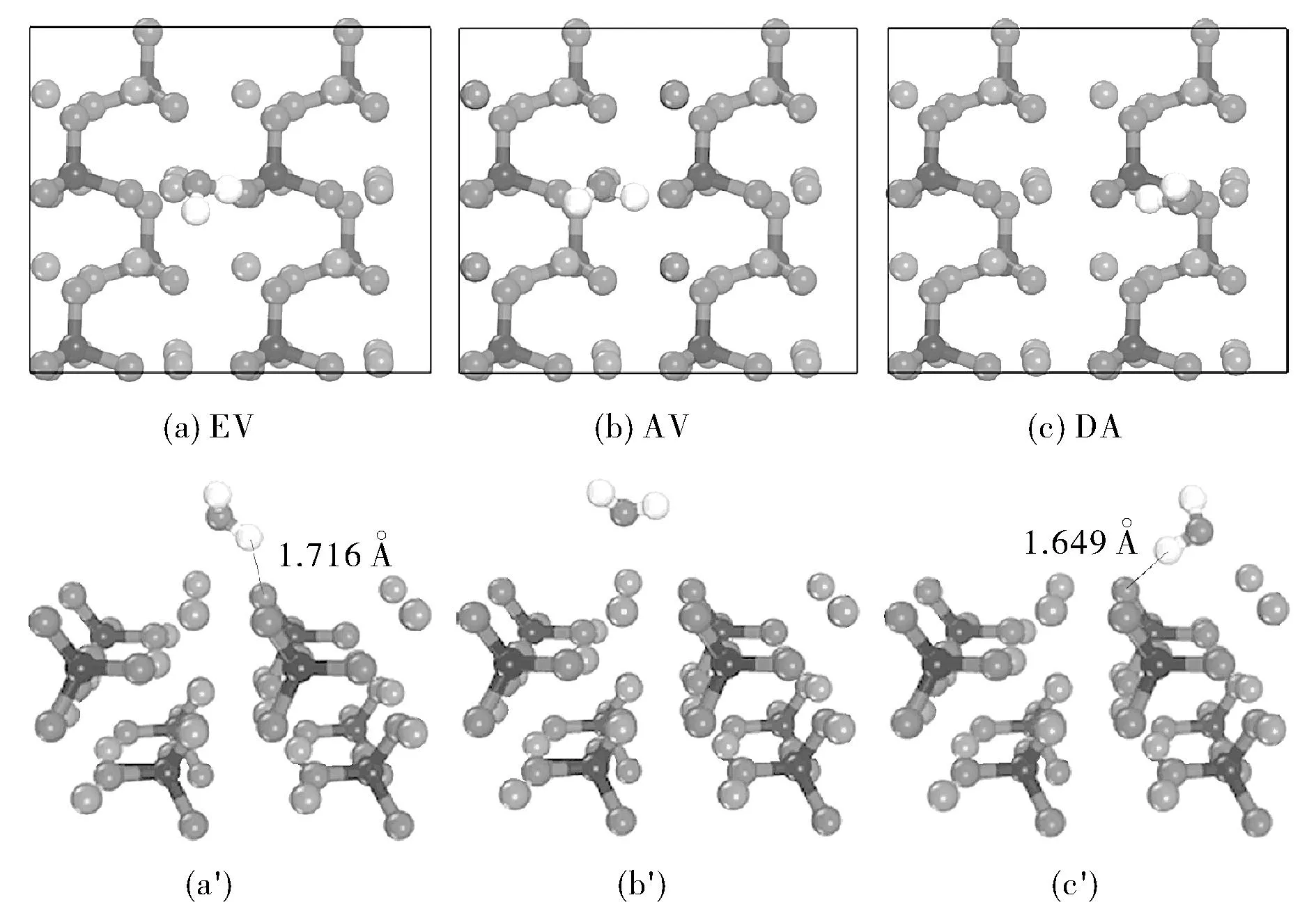
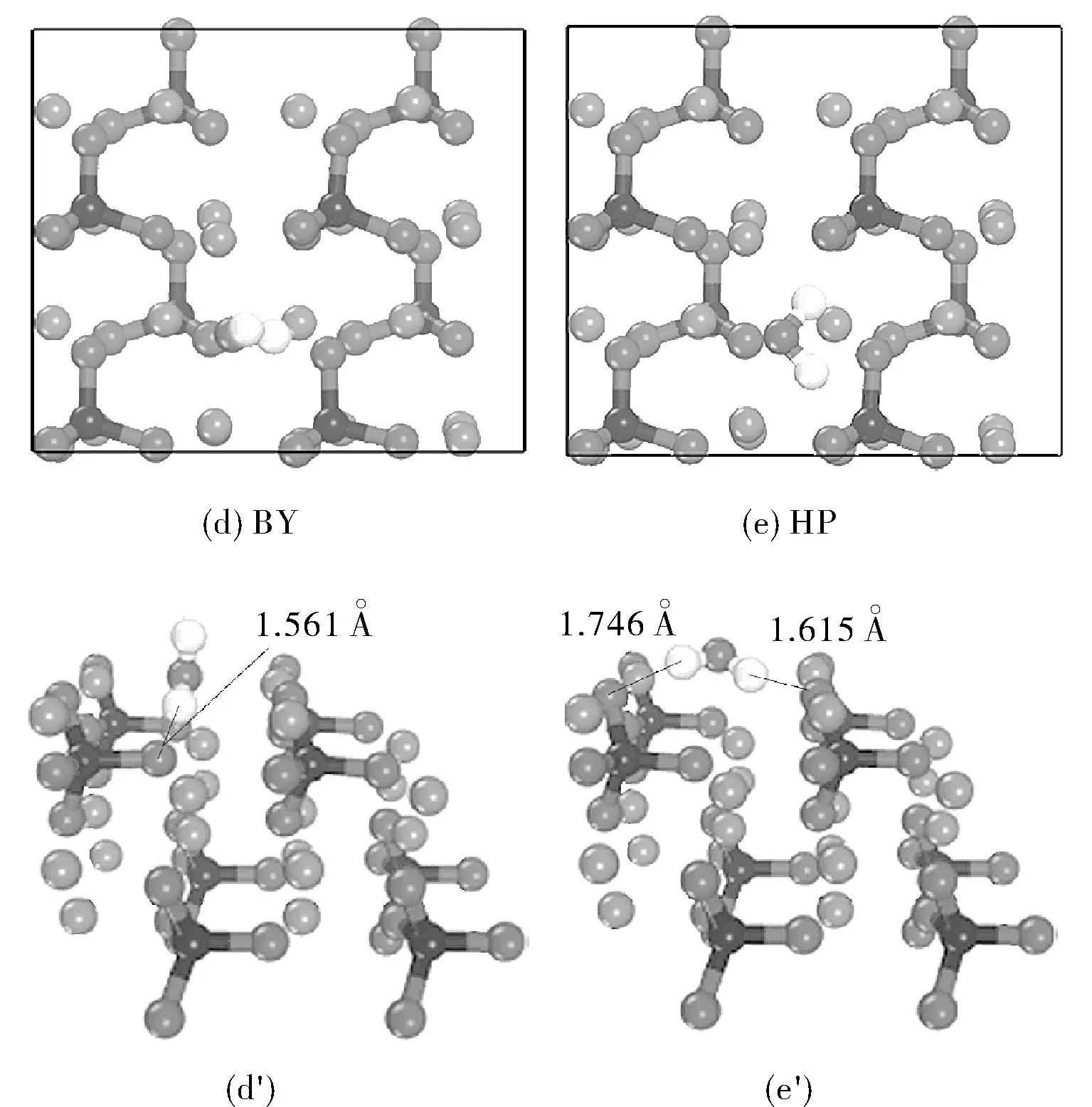
图2 水吸附在Li3PO4(100)表面的五种稳定结构的俯视图和侧视图Fig.2 Side and top views of optimized geometries for five stable configurations of water molecule adsorbed on the Li3PO4(100) surface
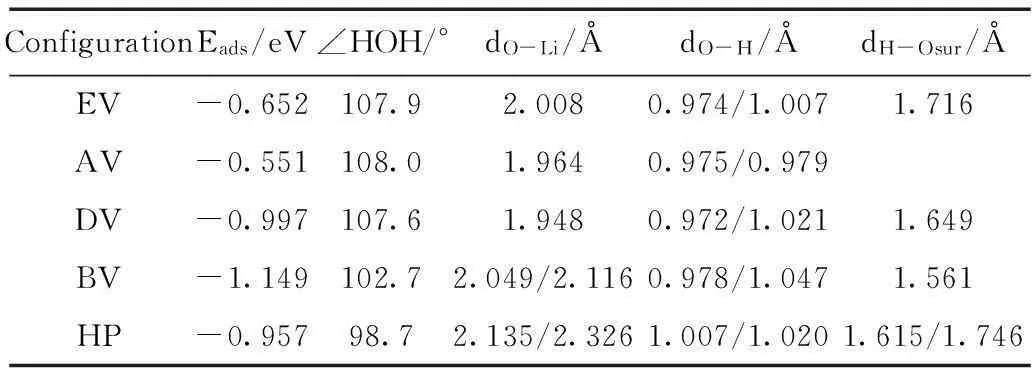
表1 水吸附在Li3PO4(100)表面优化的吸附能和结构参数Talbe 1 Adsorption energies and optimized structural parameters for water molecule adsorbed on the Li3PO4(100) surface
包括7种可能的吸附位点:(a)Li顶位,(b)O顶位,(c)Li-Li桥位,(d)Li-O长桥位, (e)Li-O短桥位, (f)O-O桥位,(g)空位。红色的球表示O原子,青色的球表示Li原子,黑色的球表示P原子。下面的图均以相同的符号表示。
dO-Li表示水分子的O原子到Li3PO4(100)表面Li原子的距离;dO-H是水分子中H原子和O原子之间的长度;dH-Osur是水中H原子到Li3PO4(100)表面Li原子的距离。
从表1可知,构型BV有着最大吸附能-1.149 eV,为最稳定的吸附构型。因此,水在Li3PO4(100)表面的最佳吸附位点为Li-Li桥位。水分子和表面基底原子间的距离分别是d(O-Li1)=2.049 Å, d(O-Li2)=2.116 Å, d(H-O)=1.561 Å。在构型EV和DV中,水分子在表面基底上,只有一个氢原子和表面基底中的O原子形成氢键,氢键的键长分别为1.716 Å和1.649 Å。然而,对于构型DV来说则是最不稳定的结构,因为该构型中不存在氢键。从构型HP中我们可以看出水分子的的取向与表面基底平行,它的两个氢原子都与表面基底中氧原子形成氢键,键长分别为1.746 Å和1.615 Å。在构型BV中,水分子中的O-H键长分别为0.978和1.047 Å,比自由水分子的略长,这主要是由于表面O原子的牵引使得羟基距离变长。在所有构型中,BV构型中形成的氢键距离最短,且水分子中的O原子同时与表面两个Li原子作用着,吸附过程中放热1.149 eV,吸附构型最稳定。
综上所述,对于水分子在Li3PO4(100)表面的吸附,水分子最容易吸附在Li-Li桥位上,其中水分子中的H原子易与表面磷酸根离子的O原子形成氢键,水分子中的O原子则被表面两个Li原子所吸引。因此,水分子在Li3PO4(100)表面之所以能稳定吸附主要取决于表面氢键的形成和水中的O原子与表面Li原子的相互作用。
2.3Mülliken布居数分析
为了进一步研究在吸附水之后水分子和磷酸锂表面之间电荷转移趋势,计算了水分子在Li3PO4(100)表面吸附之后最稳定的吸附构型BV的Mülliken电荷。计算的结果包括水分子吸附前后,水分子中的H原子和O原子,磷酸锂各个表面基底的Li原子、O原子和P原子以及水分子的电荷数都列于表2中。

表2 水吸附在Li3PO4(100)表面最稳定的构型BV的Mulliken电荷Table 2 Mulliken charges for water molecule adsorbed on the Li3PO4(100) surface of the configuration BV
从表2可以看出,在吸附水之后,水分子中的两个H原子的Mülliken电荷数值从0.53分别减少至0.47和0.42,说明水中H原子都得到电子,且靠近表面氧原子的H原子得到更多的电子;水分子中的O原子的电荷数值从-1.06增加到-1.04,说明水分子中的O原子失去了电子。Li3PO4基底表面的Li原子电荷数值从0.96增加至0.98,O原子的电荷数从-1.25增加到-1.18,这表明Li3PO4(100)表面的Li原子和O原子都失去电子。此外,从总的电荷数来看,水分子的电荷数从0.00减少到-0.15,说明水分子总体是得到电子的。整个过程P原子电荷数不变,说明P原子并没有参与电子转移。因此,总结上述可知,在构型BV中,水分子从Li3PO4(100)表面Li原子和O原子得到部分电子。
2.4态密度分析
为了更详细地研究水分子和Li3PO4(100)表面的相互作用,也计算了水分子在各个表面吸附的最稳定构型BV的分波态密度(PDOS)。图3分别给出了构型BV中的水中的H原子的1s态和基底表面磷酸根离子中的O原子的2p态以及水中O原子的2p态和基底表面Li原子的1s态的PDOS。
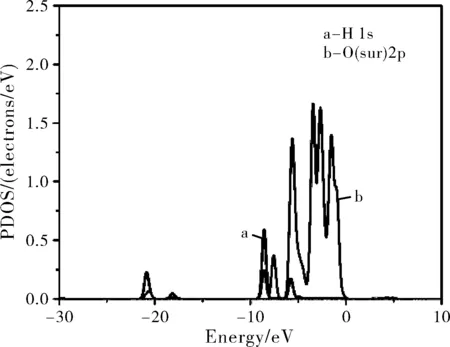
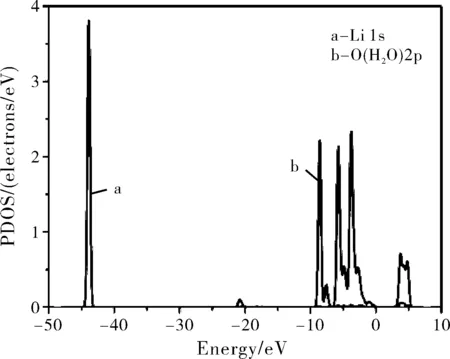
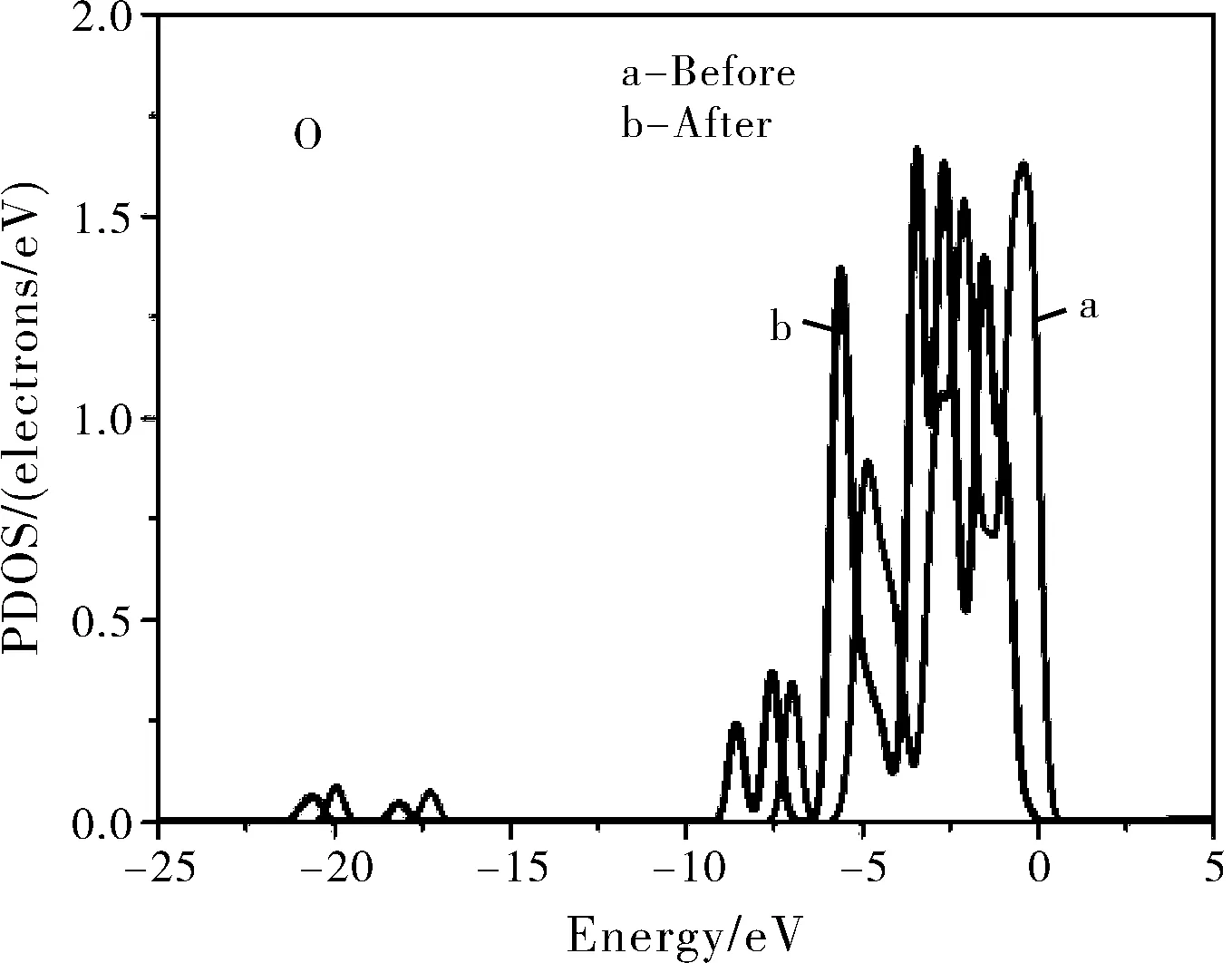
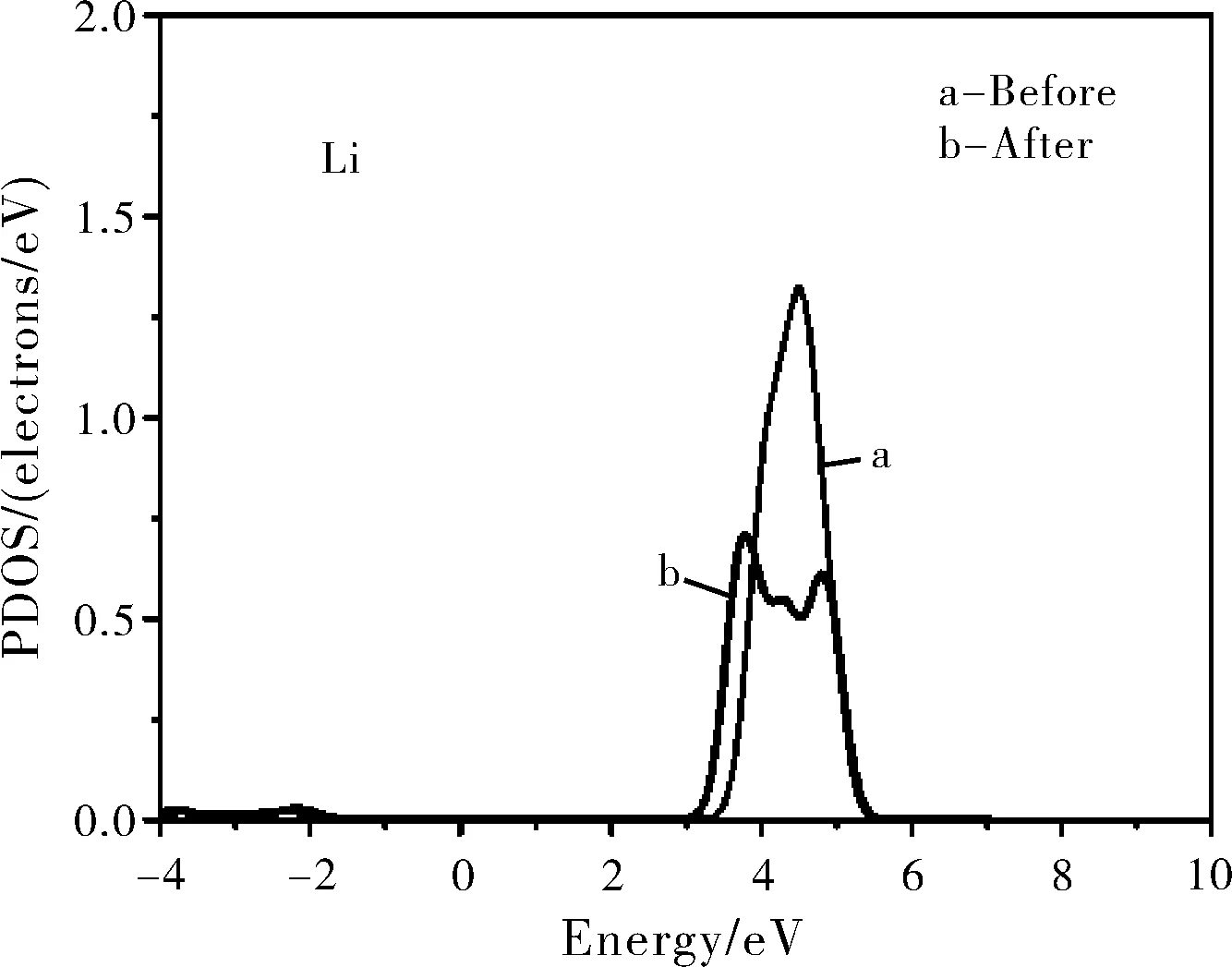
图3 水吸附在Li3PO4(100)表面最稳定的构型BV中的H2O、O和Li原子的分波态密度图(PDOS,费米能级设置在0点)Fig.3 The PDOS of water molecule, O and Li atoms for the configuration BV(Zero energy is the Fermi level)
从图3可以看出,水分子中的H 1s态和表面磷酸根离子中的O 2p态之间有三个主要的共振峰,分别为-21.5 eV、-8.6 eV和-5.7 eV,表明水分子中的H原子和Li3PO4(100)表面的O原子之间存在着相互作用,这与前面Mülliken电荷分析的结果相一致,相互作用使得水分子中的O-H键伸长(构型BV中的O-H1长1.047 Å,自由水分子中的O-H1长0.976 Å)。水分子中的O 2p态和磷酸锂表层Li 1s态的共振峰范围为-7.5~7.0 eV,表明水分子中的O原子和Li3PO4(100)表面的Li原子之间也存在着相互作用。但是水分子中的O 2p态和表面Li 1s态之间共振峰的强度很弱且重叠很小,因此判定水分子中的O原子与磷酸锂表面的Li原子之间的相互作用较弱,水分子吸附在磷酸锂表面主要依靠H原子和磷酸根离子中的O原子形成的氢键。对吸附水前后磷酸锂表面的O原子和Li原子也做了计算,发现在吸附水之后表面基底的O原子的PDOS整体向低能量处移动,有利于氢键的形成。在吸附水之后,Li原子的PDOS仅有着细微的变化,表明O-Li之间的相互作用对能隙改变的影响是非常小的。
3 结 论
本章应用密度泛函理论研究水分子在Li3PO4(100)表面的吸附。分别计算了水分子在Li3PO4(100)表面各吸附位点吸附后的几何构型和吸附能等参数。计算结果表明,水分子能自发地吸附在磷酸锂表面,水分子吸附在磷酸锂表面主要以O原子与表面Li原子靠近,最终吸附在两个Li原子形成的桥位上;而H原子与表面磷酸根离子中的O原子接触形成氢键。Mülliken布居分析结果为水分子的电荷数减少,而磷酸锂表面的Li原子和O原子电荷数增加,表明水分子从磷酸锂表面得到电子。分析分波态密度(PDOS)可得出水分子的H原子的1s轨道与表面磷酸根离子的O原子2p轨道存在较强的相互作用,O原子的2p态与表面Li原子的1s态存在相互作用且作用较弱,可以得出水分子吸附在Li3PO4(100)表面主要依靠H与表面O形成的氢键。
[2]牛锛, 满丽莹,齐恩磊,等. 磷酸锂粉体的制备与表征[J]. 硫磷设计与粉体工程, 2011(2): 27-28.
[3]Zhao S-X, Ding H, Wang Y-C, et al. Improving rate performance of LiFePO4 cathode materials by hybrid coating of nano-Li3PO4and carbon[J]. Journal of Alloys and Compounds, 2013, 566: 206-211.
[4]胡意,艾常春,刘洋. 紊流循环法合成超细磷酸锂及表征[J]. 化工学报, 2014(03):1099-1003.
[5]Mascaraque N, Tricot G, Revel B, et al. Nitrogen and fluorine anionic substitution in lithium phosphate glasses[J]. Solid State Ionics, 2014, 254: 40-47.
[6]Holzwarth N, Lepley N, Du Y A. Computer modeling of lithium phosphate and thiophosphate electrolyte materials[J]. Journal of Power Sources, 2011, 196(16): 6870-6876.
[7]Du Y A, Holzwarth N. Effects of O vacancies and N or Si substitutions on Li+migration in Li3PO4electrolytes from first principles[J]. Physical Review B, 2008, 78(17): 174301.
[8]Du Y A, Holzwarth N. First-principles study of LiPON and related solid electrolytes[J]. Physical Review B, 2010, 81(18): 184106.
[9]Filhol J S, Neurock M. Elucidation of the electrochemical activation of water over Pd by first principles[J]. Angewandte Chemie, 2006, 118(3): 416-420.
[10]Gokhale A A, Dumesic J A, Mavrikakis M. On the mechanism of low-temperature water gas shift reaction on copper[J]. Journal of the American Chemical Society, 2008, 130(4): 1402-1414.
[11]Phatak A A, Delgass W N, Ribeiro F H, et al. Density functional theory comparison of water dissociation steps on Cu, Au, Ni, Pd, and Pt[J]. The Journal of Physical Chemistry C, 2009, 113(17): 7269-7276.
[12]Jacobs G, Das T K, Patterson P M, et al. Fischer-Tropsch synthesis XAFS: XAFS studies of the effect of water on a Pt-promoted Co/Al2O3catalyst[J]. Applied Catalysis A: General, 2003, 247(2): 335-343.
[13]Xu X L, Li J Q. DFT studies on H2O adsorption and its effect on CO oxidation over spinel Co3O4(100) surface[J]. Surface Science, 2011, 605(23): 1962-1967.
[14]Cabrera-Sanfelix P, Arnau A, Darling G R, et al. Water adsorption and diffusion on NaCl (100)[J]. The Journal of Physical Chemistry B, 2006, 100(48): 24559-24564.
[15]Li B, Michaelides A, Scheffler M. Density functional theory study of flat and stepped NaCl (001)[J]. Physical Review B, 2007, 76(7): 075401.
[16]Luo J H, Zhang Y H, Li Z S. Adsorption of Water on an MgSO4(100) Surface: A First-Principles Investigation[J]. Chemphyschem, 2013, 14(9): 1969-1976.
[17]Segall M, Lindan P J, Probert M A, et al. First-principles simulation: ideas, illustrations and the CASTEP code[J]. Journal of Physics: Condensed Matter, 2002, 14(11): 2717.
[18]Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Physical review letters, 1996, 77(18): 3865-3868.
[19]Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Physical Review B, 1990, 41(11): 7892-7895.
[20]Laasonen K, Pasquarello A, Car R, et al. Car-Parrinello molecular dynamics with Vanderbilt ultrasoft pseudopotentials[J]. Physical Review B, 1993, 47(16): 10142.
[21]Keffer C, Mighell A D, Mauer F, et al. Crystal structure of twinned low-temperature lithium phosphate[J]. Inorganic Chemistry, 1967, 6(1): 119-125.
[22]Meyer H, Entel P, Hafner J. Physisorption of water on salt surfaces[J]. Surface Science, 2001, 488(1): 177-192.
Adsorption of Water on Li3PO4(100) Surface from Density Functional Theory*
LV Yan, WANG Tao, MA Wei-hua
(School of Chemical Engineering, Nanjing University of Science and Technology,JiangsuNanjing210094,China)
Lithium phosphate is an efficient catalyst for isomerization of propylene oxide. But we recently found that during the experiment after adsorbing water, the catalytic performance can be improved greatly. In order to find out what happened after adsorbing water, density functional theory (DFT) with the generalized gradient approximation (GGA-PBE) was used to study the adsorption of H2O molecule on Li3PO4(100) surface at different sites. The calculated results indicated that water molecule was inclined to locate on the Li-Li bridge site with its H atom being attracted by the O atom of phosphate ion and its O atom adjacent to two Li ions when different adsorption energies and geometrical parameters were compared. Mulliken population analysis results showed that some electrons transfered from the substrate to water molecule.
density functional theory; water molecule; Li3PO4; adsorption
国家自然科学基金面上项目(No: 21276127)。
吕岩(1991-),男,化学工程专业硕士研究生。
马卫华(1971-),女,主要从事Li3PO4催化剂的研究。
O641
A
1001-9677(2016)09-0001-04
