钇和柠檬酸对非负载磷化镍催化剂加氢脱硫性能的影响
2016-08-31徐晓伟宋华林陈彦广
宋 华, 于 祺, 徐晓伟,3, 宋华林, 姜 楠, 李 锋, 陈彦广
(1. 东北石油大学化学化工学院, 2. 石油与天然气化工省重点实验室, 大庆 163318;3. 山东玉皇化工有限公司技术与研发中心, 菏泽 274500;4. 牡丹江医学院黑龙江省高校肿瘤疾病防治重点实验室, 牡丹江 157011)
钇和柠檬酸对非负载磷化镍催化剂加氢脱硫性能的影响
宋华1,2, 于祺1, 徐晓伟1,3, 宋华林4, 姜楠1, 李锋1, 陈彦广1
(1. 东北石油大学化学化工学院, 2. 石油与天然气化工省重点实验室, 大庆 163318;3. 山东玉皇化工有限公司技术与研发中心, 菏泽 274500;4. 牡丹江医学院黑龙江省高校肿瘤疾病防治重点实验室, 牡丹江 157011)
采用传统程序升温(TPR)法制备了稀土金属钇(Y)或柠檬酸(CA)改性及Y和CA同时改性的非负载型Ni2P催化剂; 采用X射线衍射(XRD)分析、N2吸附比表面积(BET)测定、CO化学吸附分析、X射线光电子能谱(XPS)和透射电子显微镜(TEM)对催化剂的结构和性质进行了表征; 并考察了Y和CA对催化剂上进行的二苯并噻吩(DBT)加氢脱硫(HDS)反应性能的影响. 结果表明,Y和CA均可促使非晶相NixPyOz向磷化镍活性相的转化, 抑制Ni5P4杂晶相的形成, 从而促进Ni2P活性相的生成; 同时能够丰富催化剂的孔道, 抑制催化剂表面磷的富集, 得到更好的孔结构、更高的活性相分散度.Y或CA以及两者同时改性后的催化剂DBT转化率均明显高于Ni-P催化剂. 各催化剂的HDS活性大小顺序为Y-Ni-P-CA>Ni-P-CA>Y-Ni-P>Ni-P. 在340 ℃, 3.0MPa, 氢油体积比700, 质量空速(WHSV)1.5h-1的条件下,Y-Ni-P-CA催化剂的DBT转化率为97%, 比Ni-P催化剂提高了约35%.
加氢脱硫; 磷化镍; 钇; 柠檬酸; 二苯并噻吩
近年来, 随着低质量、重质化原油的逐渐增多和环境污染的不断加剧, 油品中硫、氮和氧含量过高成为环境污染的重要问题, 环保法规对燃料油中硫含量的限制越来越严格, 生产清洁燃油是炼油行业发展的必然趋势[1]. 目前, 国内外主要采用加氢脱硫(HDS)技术改善油品质量, 生产清洁燃料, 但传统的商用HDS催化剂已难以满足深度及超深度脱硫的要求[2,3]. 因此, 研发新型的高效率、高活性的深度及超深度HDS催化剂已成为从根本上解决脱硫问题的焦点[4].
过渡金属磷化物作为一类近年来发现的具备高加氢脱硫活性的催化加氢精制新材料[5~7], 与目前使用的工业催化剂相比具有更好的加氢选择性, 且耗氢量少, 因此成为加氢精制催化领域研究的热点.Oyama等[6]以二苯并噻吩(DBT)为探针, 发现过渡金属磷化物HDS和加氢脱氮活性顺序为Ni2P>WP>MoP>CoP>Fe2P.Ni2P是过渡金属磷化物中加氢脱硫性能最好的催化剂, 且具有熔点高、耐腐蚀、抗硫中毒性能高、热稳定性好及活性中心分散度高等优点. 随着环保法规提出的低硫化、无硫化趋势,Ni2P催化剂作为高效率、高活性的HDS催化剂, 将最有可能替代目前工业上使用的硫化物催化剂.
HDS催化剂中常添加各种助剂以改善其催化性能. 近年来向Ni2PHDS催化剂中添加助剂以提高其催化性能方面的研究备受关注.Li等[8]向非负载Ni2P催化剂中引入稀土金属Ce, 发现CeO2可以显著降低Ni2P的晶粒尺寸, 提高活性相分散性, 增大比表面积, 进而提高催化剂的HDS活性. 本研究组[9]制备了稀土钇(Y)改性的非负载型Ni2P催化剂, 发现引入Y可以促进非晶相NixPyOz向磷化镍活性相的转化, 在抑制Ni5P4杂晶相形成的同时, 可促进小尺寸、高分散度的Ni2P相的生成, 还可抑制催化剂表面磷富集现象, 暴露更多的金属活性位, 改善了二苯并噻吩(DBT)的HDS活性.Wang等[10]将柠檬酸(CA)引入非负载型Ni2P催化剂, 发现CA能减小Ni2P相的晶型尺寸, 从而提高催化剂的比表面积.
本文采用传统程序升温(TPR)法制备了稀土金属钇(Y)或柠檬酸(CA)改性及Y和CA同时改性的非负载型Ni2P催化剂. 采用连续流动式固定床高压微型反应装置, 以1%(质量分数)二苯并噻吩(DBT)/十氢萘溶液为模型化合物, 研究了Y和CA改性对非负载型Ni2P催化剂结构以及加氢脱硫性能的影响.
1 实验部分
1.1试剂与仪器
硝酸镍(分析纯, 北京双环化学试剂厂); 磷酸氢二铵和十氢萘(分析纯, 北京化工厂); 硝酸(分析纯, 哈尔滨市化工试剂厂); 硝酸钇和二苯并噻吩(分析纯,AladdinIndustrialCo.Ltd.); 柠檬酸(分析纯, 沈阳新兴试剂厂); 正十二烷(分析纯, 成都市科龙化工试剂厂).
X射线衍射(XRD)分析在日本理学公司D/max-2200PC型X射线衍射仪上进行, 采用CuKα辐射, 管电压40kV, 管电流30mA, 扫描速率10°/min, 扫描范围10°~80°; 催化剂比表面积(BET)采用美国Quantachrome公司NOVA2000e型测定仪, 利用低温(-196 ℃)氮气吸附法测定, 样品首先在180 ℃, 1.3kPa条件下预处理1h; 样品CO吸附表征在美国Quantachrome公司Autosorb-1-C型化学吸附仪上完成, 热导检测(TCD), 采用脉动法, 以He气作为载气. 测试前, 将催化剂在30mL/min的氢气吹扫条件下, 以10 ℃/min速率升温至500 ℃, 恒温60min;X射线光电子能谱(XPS)分析采用英国ThermoVGScientific公司的(SigmaProbe)电子能谱仪,MgKα为激发源, 电子结合能数值用样品污染碳(C1s=284.6eV)为内标进行校正; 透射电子显微镜(TEM)分析在日本电子公司JEM-1010型透射电子显微镜上进行, 加速电压100kV, 点分辨率0.4nm.
1.2实验过程
1.2.1催化剂的制备将适量的硝酸镍和磷酸氢二铵溶解于一定量的蒸馏水中(初始Ni/P摩尔比为1∶1), 搅拌得到悬浊液, 向其中加入浓硝酸, 得到澄清溶液. 将水蒸干后, 固体产物于120 ℃干燥12h, 于500 ℃焙烧3h得到磷化镍前驱体(PNi2P). 将PNi2P用氢气(200mL/min)还原, 以2.5 ℃/min的速率从室温升温至400 ℃, 再以1 ℃/min的速率升温至500 ℃, 保持2h, 然后降温至60 ℃, 氮气氛钝化处理2h(氮气流速为50mL/min), 得到的催化剂记为Ni-P.
向PNi2P前驱体中加入一定量硝酸钇溶液(Y/Ni摩尔比为0.06∶1), 搅拌30min, 浸渍12h后, 干燥, 焙烧得到Y改性的磷化镍前驱体. 还原条件与Ni-P催化剂相同, 还原后的催化剂记为Y-Ni-P.
将适量的硝酸镍和磷酸氢二铵溶解于一定量的蒸馏水中, 搅拌得到悬浊液, 向其中加入CA(CA/Ni摩尔比为2∶1), 得到澄清溶液. 干燥, 焙烧得到CA改性的磷化镍前驱体(PCANi2P). 还原条件与Ni-P催化剂相同, 还原后的催化剂记为Ni-P-CA.
向PCANi2P前驱体中加入一定量硝酸钇溶液(Y/Ni摩尔比为0.06∶1), 搅拌30min, 浸渍12h后, 干燥, 焙烧得到CA和Y同时改性的的磷化镍前驱体. 还原条件与Ni-P催化剂相同, 还原后的催化剂记为Y-Ni-P-CA.
1.2.2催化剂活性评价采用连续固定床高压微反装置进行HDS性能的评价. 反应前, 将催化剂在500 ℃下用氢气(80mL/min)预处理2h. 反应条件为340 ℃, 3.0MPa, 氢油体积比700, 质量空速(WHSV)1.5h-1. 液体产物采用配备火焰离子检测器的GC-14C型气相色谱仪(日本岛津公司)进行分析. 反应模型化合物为二苯并噻吩、十二烷烃(内标物)和十氢萘的混合溶液, 其质量分数分别为1%, 1%和98%.
2 结果与讨论
2.1XRD分析
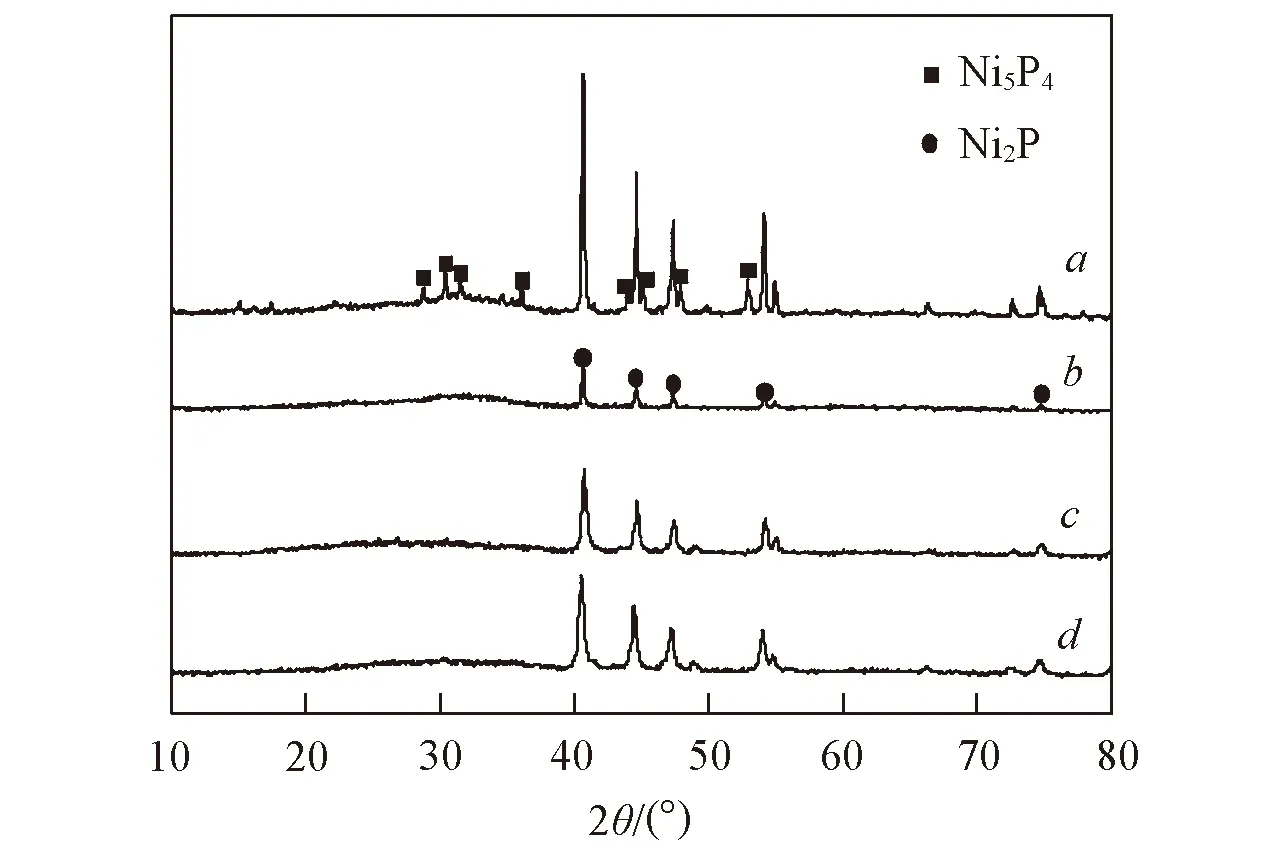
Fig.1 XRD patterns of the Ni-P(a), Y-Ni-P(b), Ni-P-CA(c) and Y-Ni-P-CA(d) catalysts
图1为Ni-P,Y-Ni-P,Ni-P-CA与Y-Ni-P-CA催化剂的XRD谱图. 由图1可知,Ni-P催化剂在2θ=40.6°, 44.5°, 47.1°, 54.1°, 54.8°, 66.1°, 72.5°和74.5°处出现了明显的Ni2P相(PDFNo. 03-0953)衍射峰, 在2θ=28.8°, 30.2°, 31.6°, 36.1°, 43.9°, 45.1°, 47.8°和53.0°处出现了较弱的Ni5P4相(PDFNo. 18-0883)衍射峰. 这表明Ni-P催化剂主要活性相为Ni2P相, 且有少量Ni5P4相. 此外,Ni-P催化剂在2θ=32°处出现较弱的弥散衍射峰, 此衍射峰为非晶相NixPyOz的衍射峰[11]. 在改性后各催化剂的XRD谱图中, 2θ=40.6°, 44.5°, 47.1°, 54.1°, 54.8°, 66.1°, 72.5°和74.5°处出现了明显的Ni2P相衍射峰, 且晶相Ni5P4和非晶相NixPyO的衍射峰基本消失, 表明单独引入Y或CA以及同时引入Y和CA均能促进非晶相NixPyOz向磷化镍活性相的转化, 同时能抑制Ni5P4杂晶相的形成, 从而促进Ni2P相的生成. 通常认为, 活性相衍射峰变宽是由于Ni2P微晶尺寸的减小. 由图1还可见, 与Ni-P相比, 改性后催化剂的衍射峰均变宽, 且衍射峰变宽大小顺序为Y-Ni-P-CA>Ni-P-CA>Y-Ni-P, 表明非负载磷化镍催化剂中Y和CA均能减小Ni2P晶粒的微晶尺寸, 促使Ni2P相更好的分散, 且Y和CA的共同作用使Y-Ni-P-CA催化剂具有更小的活性相尺寸.
2.2BET分析
表1列出了Ni-P,Y-Ni-P,Ni-P-CA与Y-Ni-P-CA催化剂的BET分析结果. 由表1可见,Ni-P催化剂的比表面积和孔容均较低, 分别为6.1m2/g和0.025cm3/g. 添加稀土Y后,Y-Ni-P催化剂的比表面积和孔容分别为12.9m2/g和0.070cm3/g, 均高于Ni-P催化剂, 且催化剂的孔径也有所提高. 这表明Y的引入改善了催化剂纳米颗粒的堆积形式, 提高了比表面积. 与Ni-P和Y-Ni-P催化剂相比, 相应的Ni-P-CA(56.5m2/g)和Y-Ni-P-CA(42.5m2/g)催化剂的比表面积分别提高了8.3和2.3倍, 同时均伴随有孔容的提高和孔径的降低. 这是由于引入CA时, 在焙烧前驱体的过程中络合物分解形成了丰富而细密的孔结构, 进而改善了催化剂的表面性质.
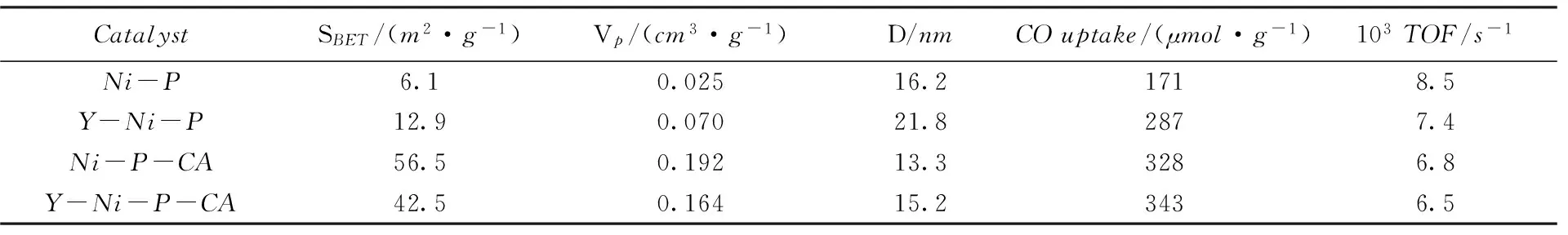
Table 1 Structural data of the Ni-P, Y-Ni-P, Ni-P-CA and Y-Ni-P-CA catalysts
2.3CO吸附结果
CO化学吸附常用于确定金属磷化物表面的金属原子数量. 通常假设在室温下每个表面Ni原子吸附1个CO分子, 同时忽略P原子对CO分子极少的吸附量,CO化学吸附可用于估算磷化镍催化剂的活性位数量及其分散度[12].Ni-P,Y-Ni-P,Ni-P-CA与Y-Ni-P-CA催化剂的CO的吸附数据见表1.Ni-P催化剂的CO化学吸附容量仅为171μmol/g. 其可能原因如下: (1)Ni-P催化剂比表面积小, 活性相分散性差; (2)催化剂表面的磷富集(见表2)覆盖了部分金属活性位. 与Ni-P催化剂相比, 改性后各催化剂的CO吸附量明显增大.Y-Ni-P和Ni-P-CA催化剂的CO吸附量分别达到287和328μmol/g,均明显高于Ni-P催化剂. 这是由于Y和CA的引入均能提高催化剂的比表面积, 改善活性相的分散性; 且对于Ni-P-CA催化剂,CA的引入在前驱体中形成络合物, 在络合物分解过程中部分磷元素损失, 致使表面P/Ni摩尔比减小(见表2), 暴露出更多的Ni金属活性位, 进而得到了较高的CO吸附容量.Y-Ni-P-CA催化剂的CO吸附量最高, 达到343μmol/g, 这是由引入的Y和CA共同作用的结果. 值得注意的是, 与Ni-P-CA催化剂相比,Y-Ni-P-CA催化剂的CO吸附量有所增加, 但增长幅度不大, 表明对于CA改性后的非负载磷化镍催化剂,Y的引入对其表面金属活性位的影响较小.

Table 2 Spectral parameters obtained by XPS analysis
2.4XPS分析

Fig.2 XPS spectra of the Ni-P(a), Y-Ni-P(b), Ni-P-CA(c) and Y-Ni-P-CA(d) catalysts (A) Ni2p core level of spectra; (B) P2p core level of spectra.
2.5TEM照片分析
各催化剂的TEM照片如图3所示, 可见,Ni-P催化剂的晶粒尺寸不均一, 这可能是由于催化剂中Ni2P和Ni5P4并存(见图1XRD分析)所致, 晶体颗粒直径约为10~15nm. 与Ni-P相比,Y-Ni-P催化剂中小颗粒的Ni5P4晶粒消失,Ni2P晶粒尺寸变化不大, 但更均一. 与Ni-P和Y-Ni-P催化剂相比, 相应的Ni-P-CA催化剂的Ni2P晶粒尺寸变化不大, 但可观察到大量小尺寸(5~7nm)的无定形Ni2P. 这些无定形Ni2P的存在是Ni-P-CA具有高比表面积的主要原因(见表1中SBET数据). 同时引入Y和CA的Y-Ni-P-CA催化剂, 基本上是由无定形Ni2P的小颗粒组成, 直径约为2~8nm, 与Ni-P-CA相比, 大颗粒的Ni2P晶粒有所减少.

Fig.3 TEM images of the Ni-P(A, A′), Y-Ni-P(B, B′), Ni-P-CA(C, C′) and Y-Ni-P-CA(D, D′) catalysts
2.6催化剂评价
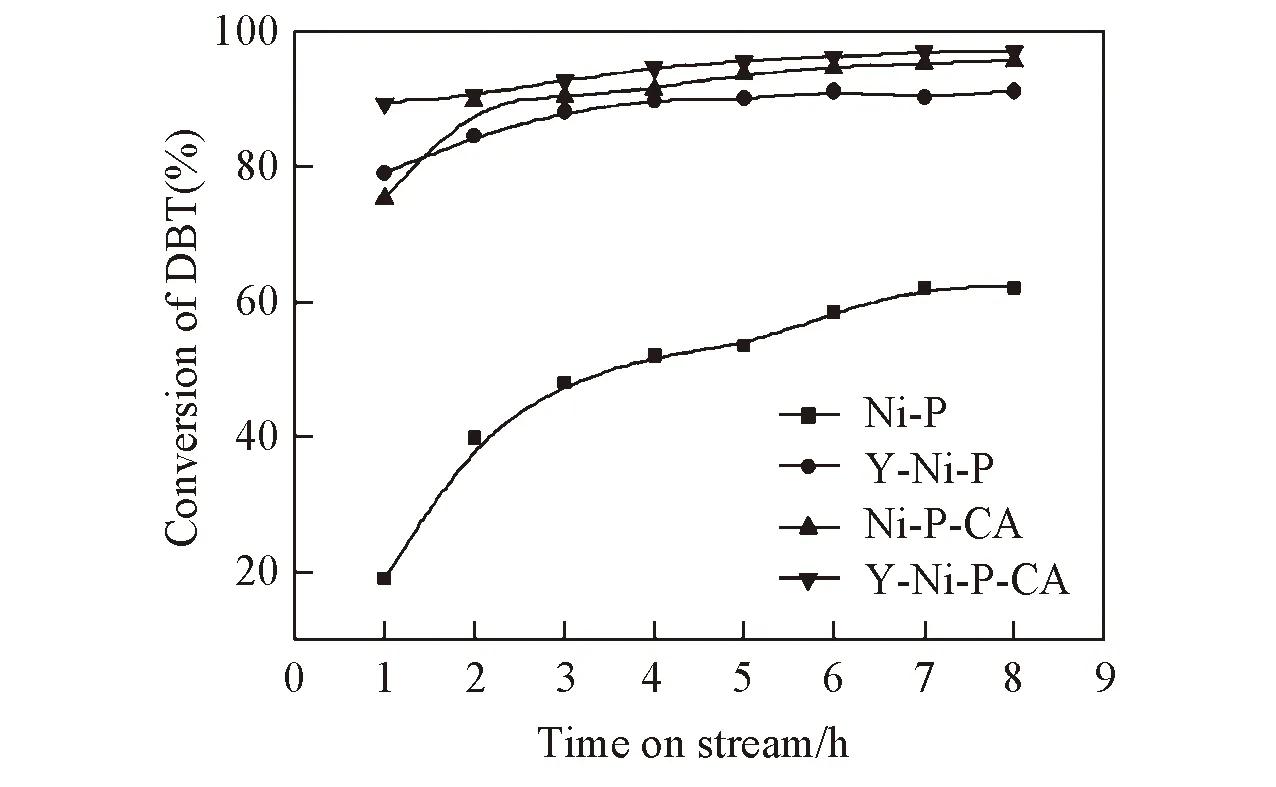
Fig.4 HDS activity of the Ni-P, Y-Ni-P, Ni-P-CA and Y-Ni-P-CA catalyst 340 ℃; 3.0 MPa; V(H2)/V(oil)=700; WHSV=1.5 h-1.
图4示出了Ni-P,Y-Ni-P,Ni-P-CA与Y-Ni-P-CA催化剂的DBT转化率随时间的变化关系. 由图4可见, 所有催化剂的HDS活性均先随反应时间延长而增大, 在反应进行一段时间后保持不变.Ni-P催化剂的DBT转化率稳定后仅为62%, 且达到稳定的时间较长. 各改性后的催化剂HDS活性达到稳定的时间明显缩短, 且DBT转化率均高于Ni-P催化剂, 表明Y和CA均对催化剂的HDS活性有重要影响.Y-Ni-P催化剂的DBT转化率为91%, 而Ni-P-CA催化剂的HDS活性为96%, 比Ni-P催化剂分别提高了29%和34%.Y和CA同时改性的Y-Ni-P-CA催化剂在不同反应时间的DBT转化率均高于Y和CA单独改性的催化剂,DBT转化率达到了97%, 与Ni-P相比提高了35%. 添加Y和CA能够提高催化剂HDS活性的原因可能是Y和CA能够抑制Ni5P4的形成(见图1中XRD照片), 从而促进Ni2P活性相的形成; 显著提高催化剂的比表面积(见表1中SBET数据), 并提高其分散度; 抑制催化剂表面磷富集, 暴露出更多Ni的活性位(见表1中CO吸附数据). 各催化剂的DBTHDS转化率大小顺序为Y-Ni-P-CA>Ni-P-CA>Y-Ni-P>Ni-P.
HDS反应稳定后, 计算各催化剂的转化频率(TOF)值, 结果如表1所示.Ni-P催化剂的TOF值为8.5×10-3s-1. 不同于CO吸附量和HDS活性的增大, 各改性后催化剂的TOF值与Ni-P催化剂相比, 均有所减小. 这可能是由于催化剂表面的金属活性位只是提供了最初的活性相, 而不是参与HDS反应的真正的活性相[14].Oyama等[6,15,16]也得到了相似的结论, 并认为在HDS反应中真正参与反应的是NixPySz活性相.
通常认为DBT的HDS反应是通过以下2个平行途径进行: (1) 直接脱硫(DDS). 该过程先生成BP, 之后BP加氢生成CHB. (2) 加氢后脱硫(HYD). 该途径先生成CHB和四氢二苯并噻吩(TH-DBT). 由于在DDS途径中BP转化为CHB的量很小, 可以忽略, 故可分别利用BP,CHB来计算催化剂DDS、HYD途径的选择性. 据文献[17]报道,Ni2P存在2种不同配位的Ni原子, 即四面体配位的Ni1位和方锥体配位的Ni2位,Ni1和Ni2均在较大的Ni2P晶粒上出现, 但Ni2位在较小的Ni2P晶粒上更多; 它们在HDS反应中分别形成了2种不同的初始活性位, 较低配位的Ni1位更有利于DDS途径脱硫, 而高配位的Ni2位则更有利于HYD途径脱硫.

Fig.5 HDS selectivity of the Ni-P, Y-Ni-P, Ni-P-CA and Y-Ni-P-CA catalysts Conditions: 340 ℃, 3.0 MPa, V(H2)/V(oil)=700, WHSV=1.5 h-1.
图5为Ni-P,Y-Ni-P,Ni-P-CA与Y-Ni-P-CA催化剂的DBTHDS产物选择性随时间的变化关系. 由图5可知, 4种催化剂的DBTHDS产物中BP的选择性均高于CHB的选择性, 再次证明在HDS反应过程中DDS途径更占优势. 与Ni-P催化剂相比,Y-Ni-P催化剂的CHB选择性稍有增大, 这可能是由于Y的引入促进了Ni2P晶粒尺寸减小, 分散度提高,Ni2位的数量增加,Ni1位数量不变, 使HDS反应中HYD途径增强的缘故;Ni-P-CA催化剂的BP选择性略有增大, 这可能是由于Y的引入抑制了催化剂表面磷富集占主导, 从而暴露出更多的Ni1位所致; 而Y-Ni-P-CA催化剂的HDS产物的选择性与Ni-P催化剂基本相同, 表明在Y和CA共同作用下, 该催化剂中参与HDS反应的Ni1位与Ni2位的比值基本不变.
3 结 论
向非负载的Ni2P催化剂中分别引入稀土金属钇(Y)、柠檬酸(CA)或同时引入Y和CA制备了系列Y,CA改性的非负载型Ni2P催化剂, 考察了Y和CA对催化剂的结构及DBTHDS性能的影响. 结果表明,Y和CA均可促使非晶相NixPyOz向磷化镍活性相的转化, 促进Ni2P活性相的生成, 同时抑制Ni5P4杂晶相的形成; 能够丰富催化剂的孔道, 显著提高催化剂的比表面积, 抑制催化剂表面磷的富集, 从而使催化剂具有更好的孔结构、更高的活性相分散度. 引入Y使HYDHDS途径增强, 提高了催化剂对CHB的选择性; 引入CA使DDSHDS途径增强, 提高了催化剂对BP的选择性;Y-Ni-P-CA催化剂的HDS产物的选择性与Ni-P催化剂基本相同, 表明在Y和CA共同作用下, 催化剂中参与HDS反应的Ni1位与Ni2位的比值基本不变.Ni-P催化剂的DBT转化率仅为62%,Y,CA改性后各催化剂DBT转化率均明显高于Ni-P催化剂.Y-Ni-P-CA催化剂的DBT转化率为97%, 比Ni-P催化剂提高了35%. 各催化剂的HDS活性大小顺序为Y-Ni-P-CA>Ni-P-CA>Y-Ni-P>Ni-P.
[1]SongH.,XuX.W.,DaiM.,SongH.L., Chem. J. Chinese Universities, 2013, 34(11), 2609—2616(宋华, 徐晓伟, 代敏, 宋华林. 高等学校化学学报, 2013, 34(11), 2609—2616)
[2]SongC.S., Catal. Today, 2003, 86(1—4), 211—263
[3]DuW.Q.,RongZ.M.,LuX.Y.,WangY.F.,LuL.H.,QuJ.P., Chem. Res. Chinese Universities, 2012, 28(5), 882—885
[4]QiheR.M.,ZhaoX.G. ,ZhangY.H. ,YuanH. ,XuG.T. , Chem. J. Chinese Universities, 2012, 33(2), 383—388(齐和日玛, 赵晓光, 张韫宏, 袁蕙, 徐广通. 高等学校化学学报, 2012, 33(2), 383—388)
[5]WangX.Q.,ClarkP.,OyamaS.T., J. Catal., 2002, 208(2), 321—331
[6]OyamaS.T., J. Catal., 2003, 216(1/2), 343—352
[7]SawhillS.J.,PhillipsD.C.,BussellM.E., J. Catal., 2003, 215(2), 208—219
[8]LiX.,ZhangY.,WangA.,WangY.,HuY., Catal. Commun., 2010, 11, 1129—1132
[9]SongH.,XuX.W.,SongH.L.,JiangN.,ZhangF.Y., Catal. Commun., 2015, 63, 52—55
[10]WangR.,SmithK.J., Appl. Catal. A, 2010, 380(1/2), 149—164
[11]SilvaV.T.,SousaL.A.,AmorimR.M.,AndriniL.,FigueroaS.J.A.,RequejoF.G.,VicentiniF.C., J. Catal., 2011, 279(1), 88—102
[12]LaymanK.A.,BussellM.E., J. Phys. Chem. B, 2004, 108, 10930—10941
[13]KorányiT.I.,VítZ.,PoduvalD.G.,RyooR.,KimH.S.,HensenE.J.M., J. Catal., 2008, 253(1), 119—131
[14]NelsonA.E.,SunM.Y.,JunaidA.S.M., J. Catal., 2006, 241(1), 180—188
[15]OyamaS.T., J. Catal., 2003, 216(1/2), 343—352
[16]OyamaS.T.,WangX.,LeeY.K.,BandoK.,RequejoF.G., J. Catal., 2002, 210(1), 207—217
[17]LiuP.,RodriguezJ.A., J. Am. Chem. Soc., 2005, 127(42), 14871—14878
(Ed.:V,Z,K)
†SupportedbytheNationalNaturalScienceFoundationofChina(No.21276048),theProjectofEducationDepartmentofHeilongjiangProvince,China(No.12541060)andtheGraduateInnovationProjectofNortheastPetroleumUniversity,China(No.YJSCX2016-019NEPU).
EffectofYttriumandCitricAcidontheHydrodesulfurizationPerformanceofUnsupportedNickelPhosphide†
SONGHua1,2,YUQi1,XUXiaowei1,3,SONGHualin4*,JIANGNan1,LIFeng1,CHENYanguang1
(1. College of Chemistry & Chemical Engineering, 2. Provincial Key Laboratory of Oil & Gas Chemical Technology, Northeast Petroleum University, Daqing 163318, China; 3. Shandong Yuhuang Chemical Co. Ltd., Heze 274500, China;4. Key Laboratory of Cancer Prevention and Treatment of Heilongjiang Province, Mudanjiang Medical University, Mudanjiang 157011, China)
TheYttrium(Y),citricacid(CA)andcombinedYandCAmodifiedbulknickelphosphide(Ni-P)catalystswerepreparedbytemperatureprogrammedreduction(TPR)method.ThecatalystswerecharacterizedbymeansofX-raydiffraction(XRD),N2-adsorptionspecificsurfaceareameasurements(BET),COchemisorptionuptakemeasurements,X-rayphotoelectronspectroscopy(XPS)andtransmissionelectronmicroscopy(TEM).TheeffectsofYandCAoncatalystactivityfordibenzothiophene(DBT)hydrodesulfurization(HDS)werestudied.TheresultsindicatedthatbothYandCAcanpromotethetransformationfromNixPyOzamorphousphasetonickelphosphideactivephase,suppresstheformationofNi5P4phaseandthereforepromotetheformationofNi2Pactivephase.AdditionofYorCAcanincreasethesurfaceareaandsuppresstheenrichmentofphosphorusonsurface,leadingtoasmallerandhighlydispersedactivephaseparticles.TheY,CAandcombinedYandCAmodifiednickelphosphidecatalystswereprovedtopossesshigherHDSactivitythanNi-Psample.TheDBTconversionsofcatalystsfollowedtheorder:Y-Ni-P-CA>Ni-P-CA>Y-Ni-P>Ni-P.Attheconditionsof340 ℃, 3.0MPa,WHSVof1.5h-1andH2/oilvolumeratioof700,theDBTconversionoftheY-Ni-P-CAcatalystreached97%,whichisanincreaseof35%whencomparedwiththatfoundforbulkNi-Psample.
Hydrodesulfurization;Nickelphosphide;Yttrium;Citricacid;Dibenzothiophene
2015-11-30. 网络出版日期: 2016-07-13.
国家自然科学基金(批准号: 21276048)、 黑龙江省教育厅面上项目(批准号: 12541060)和东北石油大学研究生创新科研项目(批准号:YJSCX2016-019NEPU)资助.
O643.361;TQ426.95
A
联系人简介: 宋华林, 男, 博士, 高级工程师, 主要从事催化剂表征方面的研究.E-mail:songhua2004@sina.com
