超高效液相色谱法同时测定肿节风中6个成分的含量
2016-08-26姚志红王其意韦卓纯龚海标姚新生
姚志红,王其意,韦卓纯,龚海标,林 培,戴 毅,2,姚新生,2
(1.暨南大学 药学院 中药及天然药物研究所,广东 广州 510632;2.中药药效物质基础及创新药物研究广东省高校重点实验室,广东 广州 510632)
超高效液相色谱法同时测定肿节风中6个成分的含量
姚志红1,2*,王其意1,韦卓纯1,龚海标1,林培1,戴毅1,2,姚新生1,2
(1.暨南大学药学院中药及天然药物研究所,广东广州510632;2.中药药效物质基础及创新药物研究广东省高校重点实验室,广东广州510632)
建立了同时测定肿节风中新绿原酸、绿原酸、隐绿原酸、咖啡酸、迷迭香酸、异嗪皮啶6个成分含量的超高效液相色谱方法。采用Waters Acquity UPLC BEH C18柱(2.1 mm×50 mm,1.7 μm)分离,以乙腈-水(各含0.1%甲酸)为流动相进行梯度洗脱,流速为0.5 mL·min-1,检测波长330 nm,柱温35 ℃;以异嗪皮啶为参照物,计算其与其余5种目标物的相对校正因子,并考察了不同色谱仪、色谱柱、流动相、流速、柱温对相对校正因子重现性的影响;通过相对校正因子计算各成分的含量,实现一测多评,同时采用外标法测定肿节风中各成分的量,并对一测多评计算值与外标法实测值进行配对t检验。肿节风中新绿原酸、绿原酸、隐绿原酸、咖啡酸、迷迭香酸、异嗪皮啶分别在0.522 0~26.09,1.482~74.13,0.591 0~29.49,0.632 5~31.29,2.612~130.5,0.970 4~48.73 μg·mL-1范围内线性关系良好(r≥0.999 7),仪器精密度、方法重复性、样品稳定性的相对标准偏差(RSD)均不大于1.9%,平均加标回收率为98.6% ~101.5%,RSD(n=6)不大于1.9%。所建立的相对校正因子重现性良好,外标法实测值与一测多评法计算值无显著性差异(P>0.05),均可用于肿节风中5个咖啡酰类和1个香豆素类成分的同时定量测定。
肿节风;超高效液相色谱法;外标法;一测多评法;定量分析;咖啡酰类成分;香豆素类成分
肿节风为金粟兰科(Chloranthaceae)植物草珊瑚(Sarcandraglabra(Thunb.)Nakai)的干燥全草,具有清热凉血、祛风通络等功效[1],用于治疗胃炎、呼吸道感染、跌打损伤等症[2-5]。肿节风中主要含有咖啡酰类、香豆素类、黄酮类、倍半萜类、有机酸类和挥发油类等成分[6],其中咖啡酰类成分如迷迭香酸等具有抗菌、抗病毒、抗炎等多种生物活性[7-8],而香豆素类成分异嗪皮啶是肿节风中的特征指标成分[1],具有抗肿瘤活性[9-10]。因此,有必要建立快速灵敏的涵盖肿节风药材多个指标和活性成分的含量测定方法。
肿节风药材多成分含量的测定多采用高效液相色谱法(HPLC),如李锦燊等[11]建立了肿节风中绿原酸、异嗪皮啶及迷迭香酸含量的HPLC测定方法,分析时长约40 min;邓伟麟等[12]建立了时长50 min的HPLC指纹图谱并测定了肿节风中异嗪皮啶与迷迭香酸的含量;吴铁荣等[13]采用HPLC建立肿节风药材中新绿原酸、绿原酸、隐绿原酸、咖啡酸、异嗪皮啶、迷迭香酸含量测定的一测多评法,但分析时长达75 min。目前采用更为高效快速的超高效液相色谱法(UPLC)开展肿节风多成分含量同时测定的研究尚未见文献报道。本课题组前期对肿节风的UPLC指纹图谱开展了研究[14],并通过对18批不同产地药材进行正交偏最小方差判别分析(OPLS-DA)发现,迷迭香酸、 新绿原酸、 咖啡酸为造成批间差异的5个主要成分中的3个已知成分,与肿节风中另两个咖啡酰类成分绿原酸和隐绿原酸均具有一定的抗炎活性[15-18],可能为肿节风发挥抗炎作用的主要活性物质群;而异嗪皮啶为肿节风中的特征指标成分。因此,对这几个成分同时开展含量测定有助于肿节风的整体质量控制。
本研究拟采用UPLC技术,利用成分内在的函数关系和比例关系[18-20],运用外标法结合一测多评法(QAMS),建立涵盖肿节风中特征指标成分以及抗炎活性成分(包括1个香豆素类成分和5个咖啡酰类成分)的含量测定方法,并借助统计学对两种方法的定量结果进行差异评价。本研究可为提高肿节风药材乃至其相关制剂的质量控制方法提供一定的实验依据。
1 实验部分
1.1仪器与试剂
美国Waters Acquity UPLC(Ultra-performance liquid chromatography) H-Class 超高效液相色谱系统,配有四元超高压溶剂系统、自动进样恒温样本管理器、柱温箱、PDA检测器和Empower 2 色谱工作站;色谱柱1为Waters Acquity UPLC BEH C18(1.7 μm,2.1 mm×50 mm);色谱柱2为Waters Acquity UPLC HSS T3(1.8 μm,2.1 mm×50 mm);色谱柱3为WatersAcquity UPLC BEH Shield RP18(1.7 μm,2.1 mm×50 mm);色谱柱4为Waters公司Acquity UPLC BEH C18(1.7 μm,2.1 mm×100 mm);微孔滤膜(孔径0.22 μm,广州市泛宏贸易有限公司);昆山KQ3200E 超声波清洗器。
乙腈(色谱纯)、超纯水(广州市信洪贸易有限公司);甲醇(色谱纯)、甲酸(分析纯)均购自山东禹王实业有限公司;屈臣氏蒸馏水(北京屈臣氏蒸馏水有限公司)。新绿原酸(批号:131229,99.58%)、绿原酸(批号:140107,99.91%)、隐绿原酸(批号:140216,98.69%)、咖啡酸(批号:140224,99.97%)均购自上海融禾医药科技发展有限公司,异嗪皮啶(萨恩化学技术(上海)有限公司,批号:BI180108,97.86%),迷迭香酸(Sigma公司,批号:MKBQ 2631V,99.22%)。
实验用18批不同产地的肿节风药材由广州市药检所中药室顾利红主任鉴定均为肿节风(Sarcandraglabra(Thunb.)Nakai)的干燥全草。
1.2色谱条件
色谱柱:Waters ACQUITY UPLC BEH C18柱(2.1 mm×50 mm,1.7 μm);流动相为乙腈(A)-水(B)(各含0.1%甲酸),梯度洗脱程序为:0~0.2 min,8%A,0.2~2.0 min,8%~12% A,2.0~7.5 min,12%~20%A,7.5~9.5 min,20%~25%A,9.5~9.6 min,25% ~8% A,9.6~11 min,8%A;检测波长:330 nm;流速:0.5 mL·min-1;柱温:35 ℃;进样量:1 μL。
1.3混合对照品溶液的配制
取新绿原酸、绿原酸、隐绿原酸、咖啡酸、异嗪皮啶、迷迭香酸6个对照品适量,分别精密称定,置于5 mL容量瓶中,用60%甲醇溶解并定容至刻度,摇匀,制成含上述6个成分分别为26.09,74.13,29.49,31.29,48.73,130.55 μg/mL的混合对照品溶液。精密吸取该混合对照品溶液,经1,4/5,3/5,1/4,1/8,1/20,1/50倍稀释成不同浓度的系列混标溶液1~7。
1.4供试品溶液的制备
取肿节风药材粉碎,过三号筛(孔径0.355 mm),取药材粉末0.5 g,精密称定,置于50 mL具塞锥形瓶中,加入60%甲醇20 mL,密塞,称定重量,超声处理(功率250 W,频率40 kHz) 30 min,取出,放冷,再称定重量。用60%甲醇补足减失的质量,摇匀,9 960 r/min离心3 min,取上清液,滤过,取续滤液,过0.22 μm滤膜,即得。
2 结果与讨论
2.1提取条件的优化
分别考察了水、60%甲醇、甲醇、70%乙醇作为提取溶剂时的超声提取率,结果发现60%甲醇较其它溶剂的提取率高,故选择60%甲醇作为提取溶剂。进一步考察了不同超声提取时间(15,30,45,60 min)的提取率,结果发现超声提取30 min时,各定量成分已提取完全,故最终选用60%甲醇超声提取30 min。
2.2色谱条件的优化
比较了乙腈-水、乙腈-水(各含0.1%甲酸)、甲醇-水和甲醇-水(各含0.1%甲酸)等流动相对肿节风药材中6个定量成分的分离情况及分析时间的影响。结果显示,流动相在不加酸的情况下,样品出峰较少,且多为宽、扁平峰,基线不稳定,影响后续的定量分析;而采用含酸的乙腈-水系统进行梯度洗脱时,各成分的分离效果较含酸的甲醇-水系统更佳,故选择乙腈-水(各含0.1% 甲酸)作为流动相。新绿原酸、绿原酸、隐绿原酸、咖啡酸和迷迭香酸为咖啡酰类成分,而异嗪皮啶为香豆素类成分,分别对这6个成分进行PDA全波长扫描,发现这两类成分在波长330 nm附近均有较好的紫外吸收,因此选择330 nm作为多成分含量测定的检测波长。在优化条件下,6种化合物混标溶液的色谱图见图1。
2.3方法学验证
2.3.1线性关系及定量下限按“1.3”方法制备不同浓度的系列混标溶液,按“1.2”色谱条件进样分析并记录上述6个成分的峰面积,以峰面积(A)为纵坐标,对应的质量浓度(C,μg·mL-1)为横坐标进行线性回归,得到肿节风药材中上述6个成分的回归方程、相关系数和线性范围。以最低浓度的对照品溶液逐级稀释,以信噪比为10对应的浓度作为定量下限(LOQ),结果见表1。6个成分在相应范围内线性关系良好,定量下限为0.122 3~0.633 4 μg/mL。

表1 肿节风中6个成分的标准曲线、线性范围及定量下限
2.3.2精密度试验取肿节风药材(批次:S7),按“1.4”方法制备供试品溶液,在优化条件下连续进样6次,测得新绿原酸、绿原酸、隐绿原酸、咖啡酸、异嗪皮啶和迷迭香酸含量的相对标准偏差(RSD)分别为0.71%,0.49%,0.52%,0.48%,0.77%,0.32%,表明仪器精密度良好。
2.3.3重复性试验取同一批肿节风药材(批次:S7),粉粹,过三号筛,分别精密称定约0.25,0.50,0.75 g,各自平行3份,按“1.4”方法制得供试品溶液,在优化条件下进行测定,测得新绿原酸、绿原酸、隐绿原酸、咖啡酸、异嗪皮啶和迷迭香酸含量的RSD值分别为1.8%,1.7%,1.9%,1.7%,0.61%,1.4%;表明该方法的重复性良好。2.3.4样品稳定性试验取同一供试品溶液,分别于室温下放置0,2,4,8,12,18,24 h,在优化条件下测得新绿原酸、绿原酸、隐绿原酸、咖啡酸、异嗪皮啶和迷迭香酸峰面积的RSD分别为1.2%,0.80%,1.2%,1.5,1.1%,0.41%。结果表明,供试品溶液在室温下24 h内稳定。2.3.5加样回收率试验取肿节风药材(批次:S7),粉粹,过三号筛,精密称定6份,每份0.25 g,分别准确加入6个对照品,加入量见表2 ,按“1.4”所述自“置于50 mL具塞锥形瓶中”起之后的操作进行加混合标准品的供试品溶液制备,在优化条件下测定,结果列于表2。测得6个成分的平均回收率为98.6%~101.5%,RSD(n=6) 不大于1.9%。本法用于肿节风6个成分含量测定的准确度良好。
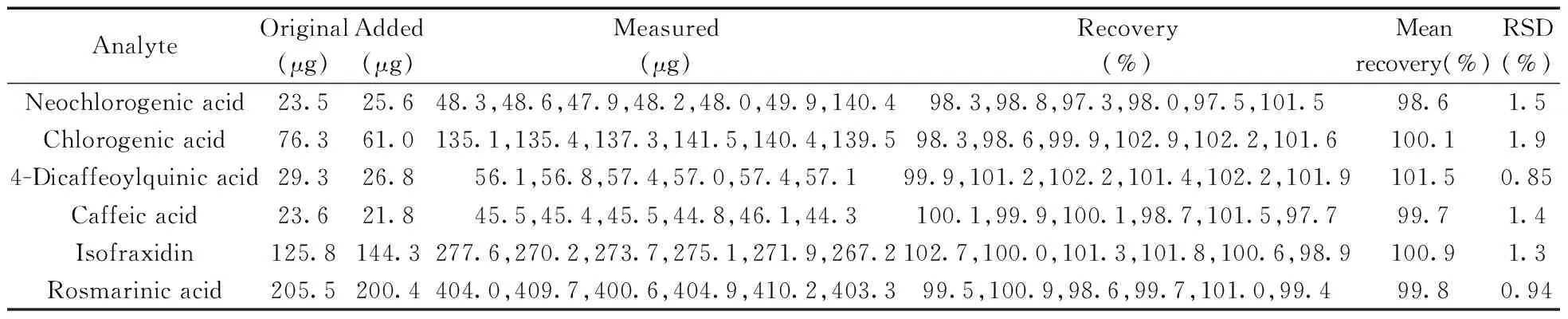
表2 肿节风中6个成分的加样回收率(n=6)
2.4相对校正因子计算及重现性考察
2.4.1QAMS法测定相对校正因子及含量一测多评法(Quantitative analysis of multi-components by single marker,QAMS)可同时进行多成分的含量计算,首先配制混标溶液和样品溶液,选择某特征定量成分s为参照物,针对混合对照品溶液结果按公式(1)计算s相对于其他定量成分i的相对校正因子RCF(即fsi),再通过样品溶液的测定结果按公式(2)计算成分i的浓度Ci。首先按“1.3”配制系列混标溶液,按“1.2”色谱条件进样测定并记录6个定量成分的峰面积,以异嗪皮啶(图1峰5)为内参,分别计算其相对于其他5个成分(图1峰1 ~4,6)的相对校正因子,按“1.4”配制样品溶液,以“1.2”色谱条件进样测定并记录6个定量成分的峰面积,根据相对校正因子计算其他5个成分(1~4,6) 的含量。
(1)
(2)
式中As为内参物对照品s的峰面积,Cs为内参物对照品s的浓度,Ai为某待测成分对照品i的峰面积,Ci为某待测成分对照品i的浓度。
2.4.2不同色谱柱与超高效液相色谱仪对相对校正因子的影响采用WatersUPLCBEHC18(2.1mm×50mm,1.7μm),WatersUPLCHSST3(2.1mm×50mm,1.8μm),WatersUPLCBEHShieldRP18(2.1mm×50mm,1.7μm)和WatersUPLCBEHC18(2.1mm×100mm,1.7μm) 4种色谱柱以及WatersAcquityUPLCH-Class和WatersAcquityUPLC两种超高效液相色谱仪对相对校正因子的重现性进行考察。精密吸取“1.3”制备的系列混标溶液,依“1.2”色谱条件分别进样测定,计算其相对校正因子,结果列于表3。结果表明不同仪器和色谱柱(1~4)测定的6个定量成分相对校正因子的RSD均不大于2.9%,重现性良好。
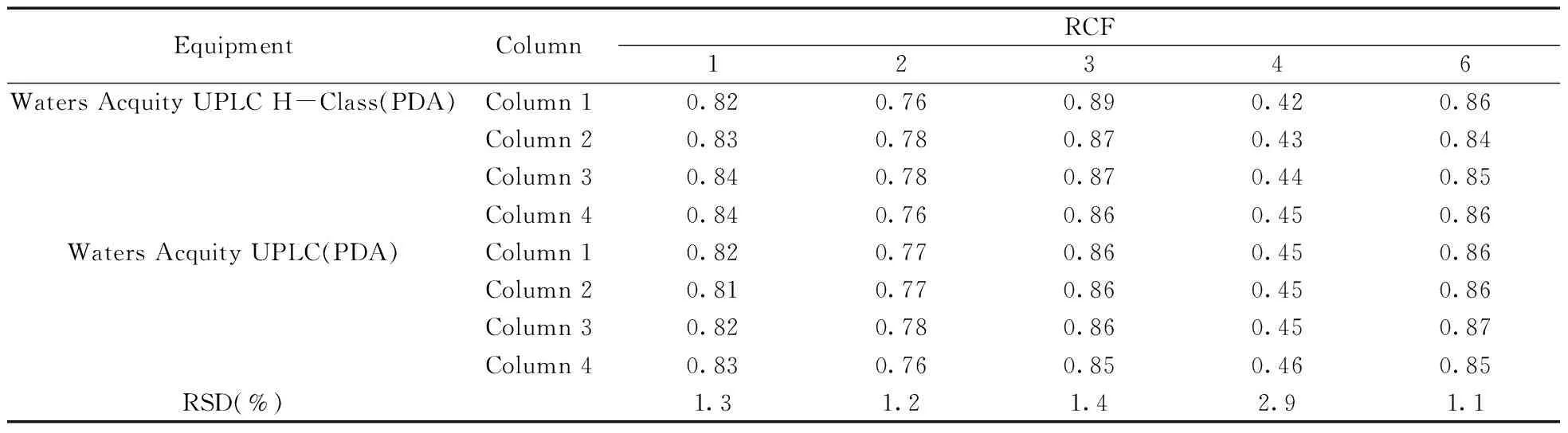
表3 肿节风中6个成分采用不同仪器和色谱柱测定的相对校正因子结果(n=7)
PDA:photo-diode array(光电二极管阵列检测器);1.neochlorogenic acid;2.chlorogenic acid;3.4-dicaffeoylquinic acid;4.caffeic acid;5.isofraxidin(internal standard);6.rosmarinic acid
2.4.3不同流动相系统、柱温、流速、进样体积对相对校正因子的影响分别采用327,330,333 nm 3个检测波长,0.4,0.5,0.6 mL·min-13种流速,30,35,40 ℃ 3种柱温和1.0,1.5,2.0 μL 3种进样体积对相对校正因子的重现性进行考察,其他色谱条件同“1.2”,精密吸取“1.3”制备的系列混标溶液,分别进样测定,计算其相对校正因子,得不同检测波长(表4)和不同流速下测定的相对校正因子的RSD均不大于1.4%;不同进样体积测定的相对校正因子的RSD值均不大于1.7%;不同柱温测定的相对校正因子的RSD值均不大于1.5%。表明本方法在上述不同实验条件下的相对校正因子重现性良好。

表4 肿节风中6个成分在不同检测波长下测定的相对校正因子结果(n=7)
the number(1-6) denoted was the same as that in Table 3
2.4.4待测组分色谱峰的定位色谱峰定位常用相对保留值(ris)和保留时间差(ΔtRis)作为定位依据。相对保留值是指各待测成分(i)与内参物(s)保留时间的比值,计算公式:ris=tRi/tRs;保留时间差是指各待测成分(i)与内参物(s)保留时间的差值,计算公式:ΔtRis=tRi-tRs。实验对Waters Acquity UPLC H-Class和Waters Acquity UPLC两种超高效液相色谱仪,Waters UPLC BEH C18(2.1 mm×50 mm,1.7 μm)、Waters UPLC HSS T3(2.1 mm×50 mm,1.8 μm)、Waters UPLC BEH Shield RP18(2.1 mm×50 mm,1.7 μm)、Waters UPLC BEH C18(2.1 mm×100 mm,1.7 μm) 4种色谱柱分别进行了相对保留值和保留时间差的重现性考察,按“1.2”色谱条件分别进样测定,结果上述6个成分相对保留值和保留时间差的RSD均大于5.0%,未能准确用于待测组分的色谱峰定位。色谱峰定位常以相对保留值和保留时间差作为评价依据,但该法仅适用于待测组分与内标保留时间差别较小的色谱峰,随着待测组分与内标保留时间差的增大,待测色谱峰的相对保留时间越易受色谱条件的影响[21]。考察了相对保留值与相对保留时间差值在不同仪器和不同色谱柱中的重现性,结果发现,随着各待测组分的保留时间远离参照峰,其相对保留时间值的RSD逐渐增大,而相对保留时间差值的波动更大,在这种情况下,利用相对保留时间值和相对保留时间差值很难准确定位,由此可见该法仅适用于相同填料或色谱行为极为相似的色谱柱。因此,为了保证待测色谱峰的准确定位,在实际应用中应对色谱柱型号和规格进行规定。此外,由考察结果看,6个成分的出峰顺序未受仪器和色谱柱型号的影响,因而也可考虑制备“对照提取物”进行色谱峰定位。所谓“对照提取物”,即指化学组成相对固定,主要药效成分有一定定量指标的中药“标准”提取物,其本身是一种混标,具有良好的均匀性、稳定性,且专属性较强,价格低,可用于色谱鉴别。目前本课题组正在开展肿节风的“对照提取物”研究,未来有望为本方法提供技术支持。
2.5外标法与一测多评法的结果比较
取18批次肿节风药材,按“1.4”方法制备供试品溶液,按“1.2”色谱条件分别进样测定,记录峰面积,分别采用外标法(ESM)测定和一测多评法(QAMS)计算6个成分的含量,结果见表5。对两种方法测定的含量进行配对t检验,结果表明,两种方法测得结果无显著性差异(P>0.05)。
表5外标法和一测多评法测得18批肿节风药材中6个不同成分的含量
Table 5Contents of six different components in 18 batches ofHebraSarcandraeby ESM and QAMS
(mg·g-1)
3 结 论
本研究采用UPLC技术,以“一测多评”法测定了肿节风中异嗪皮啶、新绿原酸、绿原酸、隐绿原酸、咖啡酸、迷迭香酸6个成分的含量,与HPLC测定上述成分所需分析时间75 min[13]相比,本方法仅用11 min即实现了6个目标成分的高效分离和快速分析,相对校正因子的重现性良好,“一测多评”法与外标法测定的结果无显著性差异,表明该方法可靠、准确,可实现只用异嗪皮啶一种对照品同时测定肿节风中6个成分的含量。
[1]Pharmacopoeia Commission of China.Pharmacopoeia of the People's Republic of China(1st vol).Beijing:China Medical Science Press(中华人民共和国药典委员会.中华人民共和国药典(第1 部).北京:中国医药科技出版社),2010:207.[2]Hou J,Li M F.ShanghaiMed.Pharm.J.(侯静,李明峰.上海医药),2009,30(6):283-285.
[3]Yao S X,Dong Y.Clin.Med.(姚圣祥,董艳.临床医学),2005,11:73-74.
[4]Lin M,Mei J,Liu X W.Mod.Med.Health(林敏,梅娇,刘兴文.现代医药卫生),2009,25(6):901-902.
[5]Li Q,Zheng L J,Hao Z H,Du M.ShanghaiMed.Pharm.J.(李晴,郑立君,郝振宏,杜民.上海医药),2008,29(9):420-421.
[6]Tong S Q,Huang J,Wang B L,Yan J Z.Chin.Tradit.HerbalDrugs(童胜强,黄娟,王冰岚,颜继忠.中草药),2010,41(2):198-201.
[7]Swarup V,Ghosh J,Ghosh S,Saxena A,Basu A.Antimicrob.AgentsChemother.,2007,51(9):3367-3370.
[8]Prasad N R,Jeyanthimala K,Ramachandran S.J.Photochem.Photobiol.B,2009,95(3):196-203.
[9]Niu X F,Xing W,Li W F,Fan T,Hu H,Li Y M.Int.Immunopharmacol.,2012,14(2):164-171.
[10]Yamazaki T,Tokiwa T.Biol.Pharm.Bull.,2010,33(10):1716-1722.
[11]Li J S,Wu H W.NorthwestPharm.J.(李锦燊,吴洪文.西北药学杂志),2014,2:125-127.
[12]Deng W L,Wu Y Y,Bi D,Duan R,Zhan H Q,Dong T X.ChinaPharm.(邓伟麟,吴莹莹,毕丹,段然,詹华强,董婷霞.中国药房),2013,47:4472-4474.
[13]Wu T R,Xu Y,Luo Y H.J.Chin.Med.Mater.(吴铁荣,许妍,罗跃华.中药材),2011,11:1730-1734.
[14]Wei Z C,Yao Z H,Wang Q Y,Lin P,Gu L H,Dai Y,Yao X S.Chin.Tradit.HerbalDrugs(韦卓纯,姚志红,王其意,林培,顾利红,戴毅,姚新生.中草药),2015,6:895-900.
[15]Da Cunha F M,Duma D,Assreuy J,Buzzi F C,Niero R,Campos M M,Calixto J B.FreeRadic.Res.,2004,38(11):1241-1253.
[16]Yang B,Qiu Y,Wang L P,Zhang X L.Chin.Pharmacol.Bull.(杨斌,丘岳,王柳萍,张锡流.中国药理学通报),2009,25(4):542-545.
[17]Song Y L,Wang H M,Ni F Y,Wang X J,Zhao Y W,Huang W Z,Wang Z Z,Xiao W.Chin.Tradit.HerbalDrugs(宋亚玲,王红梅,倪付勇,王雪晶,赵祎武,黄文哲,王振中,萧伟.中草药),2015,4:490-495.
[18]Wang Z M,Gao H M,Fu X T,Wang W H.ChinaJ.Chin.Mater.Med.(王智民,高慧敏,付雪涛,王维皓.中国中药杂志),2006,31(23):1925-1928.
[19]Wang R,Huang S J,Wang Z T.J.ShenyangPharm.Univ.(王瑞,黄山君,王峥涛.沈阳药科大学学报),2011,28(8):594-598.
[20]Wu D,Zang Z L,Wang D Q,Li C Y.Chin.Pharm.J.(吴笛,臧忠良,王德勤,李楚源.中国药学杂志),2012,47(18):1509-1513.
[21]Wang L X,Xiao H B,Liang X M.Chin.J.Anal.Chem.(王龙星,肖红斌,梁鑫淼.分析化学),2003,10:1232-1236.
Quantitative Analysis of 6 Components in Hebra Sarcandrae by Ultra Performance Liquid Chromatography
YAO Zhi-hong1,2*,WANG Qi-yi1,WEI Zhuo-chun1,GONG Hai-biao1,LIN Pei1,DAI Yi1,2,YAO Xin-sheng1,2
(1.Institute of Chinese Materia Medica & Natural Products,College of Pharmacy,Jinan University,Guangzhou510632,China;2.Guangdong Province Key Laboratory of Pharmacodynamic Constituents of TCM and New DrugResearch,Guangzhou510632,China)
An ultra performance liquid chromatographic(UPLC) method was developed to quantitatively analyze six components,i.e.,neochlorogenic,chlorogenic acid,4-dicaffeoylquinic acid,caffeic acid,rosmarinic acid and isofraxidin inHebraSarcandrae.The UPLC analysis was carried out on a Waters Acquity BEH C18column(2.1 mm×50 mm,1.7 μm) by gradient elution with acetonitrile and water(both containing 0.1% formic acid) as mobile phase at a flow rate of 0.5 mL·min-1.The column temperature was maintained at 35 ℃ and the detection wavelength was set at 330 nm.Isofraxidin was used as an internal reference substance to get the average relative correction factors(RCFs) of the other 5 components(neochlorogenic,chlorogenic acid,4-dicaffeoylquinic acid,caffeic acid,rosmarinic acid),and chromatographic column,mobile phase,flow rate,column temperature were investigated to validate the durability of the achieved RCFs.The contents of 6 components were determined by both quantitative analysis of multicomponents by single marker(QAMS) and external standard method(ESM),and the quantitative results were compared between the two methods by using pairedt-test.The results showed that the linear ranges for neochlorogenic,chlorogenic acid,4-dicaffeoylquinic acid,caffeic acid and,rosmarinic acid and isofraxidin were 0.522 0-26.09,1.482-74.13,0.591 0-29.49,0.632 5-31.29,2.612-130.5,0.970 4-48.73 μg·mL-1(r≥0.999 7),respectively.The RSD values(n=6) of precision,reproducibility,and sample stability were not more than 1.9%.The average recoveries of 6 components were between 98.6% and 101.5% with RSDs not more than 1.9%.The achieved RCFs are good(RSD<5.0%).There is no significant difference in the contents obtained by the two methods withPvalues greater than 0.05,so both of the methods could be applied in the simultaneous determination of five caffeoyl derivatives and one coumarin inHebraSarcandrae.
HebraSarcandrae;ultra performance liquid chromatography(UPLC);external standard method;quantitative analysis of multicomponents by single marker;quantitative analysis;caffeoyl derivatives;coumarin
2015-12-07;
2016-01-05
姚志红,副教授,研究方向:中药质量控制、药物代谢及动力学研究,Tel:020-85221767,E-mail:tyaozh@jnu.edu.cn
doi:10.3969/j.issn.1004-4957.2016.06.002
O657.72;TQ460.72
A
1004-4957(2016)06-0641-07
