化妆品中致敏原香豆素及其衍生物的高效液相色谱法测定及质谱确证
2016-08-19孟宪双袁汉成郭项雨
孟宪双,马 强*,袁汉成,白 桦,张 庆,郭项雨
(1.中国检验检疫科学研究院,北京 100176;2.沃特世科技(上海)有限公司,上海 201206)
化妆品中致敏原香豆素及其衍生物的高效液相色谱法测定及质谱确证
孟宪双1,马强1*,袁汉成2,白桦1,张庆1,郭项雨1
(1.中国检验检疫科学研究院,北京100176;2.沃特世科技(上海)有限公司,上海201206)
建立了化妆品中致敏原香豆素及其7种衍生物(二氢香豆素、7-甲氧基香豆素、7-甲基香豆素、7-乙氧基-4-甲基香豆素、环香豆素、双香豆素和醋硝香豆素)的高效液相色谱测定方法。样品以0.1 mol/L氢氧化钠溶液-乙腈(1∶9,体积比)混合溶液进行超声提取,提取液经高速离心及微孔滤膜过滤后,ZORBAX SB-C18(250 mm×4.6 mm,5 μm)色谱柱分离,0.05 mol/L乙酸铵溶液(含0.25%乙酸)和乙腈为流动相梯度洗脱,二极管阵列检测器检测,外标法定量。疑似阳性样品采用高效液相色谱-串联质谱法进行确证。结果表明,香豆素及其衍生物在一定浓度范围内线性良好(r2≥0.999 6),定量下限为8~20 mg/kg。在低、中、高3个加标水平下,香豆素及其衍生物的平均回收率为84.6%~100.7%,相对标准偏差(n=6)为0.8%~9.7%。该方法准确、稳定性好、特异性强,能够为化妆品检验和日常生产质量控制提供科学依据及技术支持。
香豆素及其衍生物;致敏原;高效液相色谱;高效液相色谱-串联质谱;化妆品
香豆素天然产物广泛分布于植物界[1],在日用化妆品中,通过添加一定量的香豆素类物质,可增强化妆品的香气[2]。香豆素类物质对各种波长的光均有较强吸收,可引起皮肤的光敏感作用[3],部分香豆素类物质可引发肝损伤[4],在使用高含量香豆素类化妆品的过程中,该类化合物可能通过皮肤进入体内[5]。欧盟化妆品指令(76/768/EEC)第七修正案规定[6],香豆素作为致敏原在即洗型化妆品中含量超过0.01%或在滞留型化妆品中含量超过0.001%时必须在化妆品标签上予以标注。鉴于香豆素衍生物可能具有与香豆素类似的毒副作用[7],我国《化妆品卫生规范》(2007年版)[8]中规定,化妆品中限制或禁止使用香豆素衍生物,其中二氢香豆素、双香豆素、环香豆素、醋硝香豆素、7-甲氧基香豆素、7-甲基香豆素、7-乙氧基-4-甲基香豆素均被列入禁用物质清单。
关于香豆素类致敏原的仪器分析方法大多为高效液相色谱法(HPLC)[9-11]、高效液相色谱-串联质谱法(HPLC-MS/MS)[12-13]、气相色谱法(GC)[14]、气相色谱-质谱法(GC-MS)[15]、气相色谱-串联质谱法(GC-MS/MS)[16-17]等,采用的前处理方法多以甲醇或乙醇为提取溶剂超声提取。考虑到甲醇对唇膏等蜡质类化妆品的分散效果不佳,不利于有效提取,并且目标物质在碱性溶剂中溶解效果较好,本研究比较了不同溶剂的提取效果,最终选用0.1 mol/L氢氧化钠溶液-乙腈(1∶9,体积比,下同)混合溶液作为提取溶剂(对于蜡质类样品首先以四氢呋喃进行分散)对香豆素类致敏原进行超声提取。优化了各目标分析物的检测波长,并针对双香豆素色谱峰形进行了流动相种类及改性剂浓度的优化,建立了化妆品中香豆素致敏原及其7种衍生物的高效液相色谱分析方法。对于疑似阳性样品,进一步采用高效液相色谱-串联质谱法进行确证,可有效避免假阳性结果产生。本方法准确、可靠、特异性强且稳定性好,可为化妆品中香豆素类致敏原的风险监测提供准确可靠的技术手段。
1 实验部分
1.1仪器与试剂
Agilent 1200高效液相色谱仪,配二极管阵列检测器(美国Agilent公司);ACQUITY超高效液相色谱仪、XEVO TQ三重四极杆质谱仪、MassLynx数据处理系统(美国Waters 公司);UV-3600型紫外可见光分光光度仪(日本Shimadzu公司);Milli-Q超纯水器(美国Millipore公司);CR 21G高速冷冻离心机(日本Hitachi公司);KQ-600超声波清洗器(昆山市超声仪器有限公司);MS2型涡旋振荡器(德国IKA公司);AB204-S电子天平(美国Mettler Toledo公司)。
香豆素(CAS 91-64-5)、二氢香豆素(CAS 119-84-6)、7-甲氧基香豆素(CAS 531-59-9)、7-甲基香豆素(CAS 2445-83-2)、7-乙氧基-4-甲基香豆素(CAS 87-05-8)、环香豆素(CAS 518-20-7)、双香豆素(CAS 66-76-2)及醋硝香豆素(CAS 152-72-7)标准品均购自德国Dr.Ehrenstorfer公司,纯度均大于98%;甲醇、乙腈、四氢呋喃及二氯甲烷为色谱纯(美国Fisher公司);乙酸铵、氢氧化钠(北京化工厂)及乙酸(国药集团化学试剂有限公司)均为分析纯。
1.2高效液相色谱测定条件
色谱柱:ZORBAX SB-C18(250 mm×4.6 mm,5 μm);流动相:A为0.05 mol/L乙酸铵(含0.25%乙酸),B为乙腈;梯度洗脱程序:0~5.0 min,70%~55% A;5.0~8.0 min,55%~50% A;8.0~8.2 min,50%~10% A;8.2~13.0 min,10% A;13.0~13.2 min,10%~5%A;13.2~16.0 min,5% A;16.0~16.2 min,5%~70% A;16.2~18.0 min,70% A;柱温:30 ℃;流速:1.0 mL/ min;进样量:10 μL;检测波长:香豆素、二氢香豆素、7-甲基香豆素和环香豆素为280 nm,双香豆素和醋硝香豆素为306 nm,7-甲氧基香豆素和7-乙氧基-4-甲基香豆素为320 nm。
1.3高效液相色谱-串联质谱确证条件
色谱柱:XBridge Phenyl(150 mm×2.1 mm,3.5 μm);流动相:A为水,B为乙腈;梯度洗脱程序:0~4.5 min,70%~35% A;4.5~5.0 min,35%~5% A;5.0~6.0 min,5%A;6.0~6.1 min,5%~70%A;6.1~8.0 min,70%A;流速:0.3 mL/min;柱温:35 ℃;进样量:5 μL。
电喷雾离子源(ESI);毛细管电压:3.0 kV;脱溶剂气:500 ℃,1 000 L/h;锥孔气流速:50 L/h;离子源温度:150 ℃;萃取电压:3.0 V;碰撞气为氩气;多反应监测(MRM)模式;香豆素及其衍生物的前体离子、产物离子、锥孔电压及碰撞能量等质谱分析参数见表1。

表1 香豆素及其衍生物的质谱分析参数
*ions with higher abundance
1.4标准储备液及标准工作液的配制
称取各标准物质10 mg(精确至0.1 mg)于10 mL容量瓶中,除双香豆素先用5 mL二氯甲烷溶解,再用乙腈定容至刻度外,其它目标物均直接以乙腈溶解并定容至刻度。使用时以乙腈逐级稀释成适宜浓度的混合标准溶液,均于4 ℃避光保存。
1.5样品预处理
1.5.1化妆水、膏霜、香波与散粉类样品称取0.5 g试样(精确至0.001 g)于具塞比色管中,加入8 mL 0.1 mol/L氢氧化钠溶液-乙腈(1∶9),涡旋振荡混匀,在超声波清洗器中超声提取30 min,以1.0 mol/L乙酸溶液调节pH值至中性,并用乙腈定容至10 mL,混匀,以10 000 r/min离心10 min,上清液过0.45 μm微孔滤膜,滤液供上机测定。
自2013年起,按照河北省文化厅边远贫困地区人才支持计划文化工作者专项工作要求与部署,河北省图书馆已圆满完成4年的人才支持项目计划,其中包括人员的选派工作和培养工作。在这四年间里,河北省图书馆共计派出业务骨干52人次进行业务指导,包括读者服务标准化、业务管理自动化、数字资源推广、文献资源建设规范化等技术指导;为国定贫困县公共图书馆培养了34名文化工作者,主要形式为组织各基层贫困县公共图书馆工作人员到省馆实地培养、以干代训,让参加培养的学员边学习、边实践,在实际工作中提高理论基础和业务技能。
1.5.2唇膏类样品称取0.5 g试样(精确至0.001 g)于具塞比色管中,先加入2 mL四氢呋喃,涡旋振荡混匀,再加入6 mL 0.1 mol/L 氢氧化钠水溶液-乙腈(1∶9),涡旋振荡混匀,其余步骤同“1.5.1”所述。
2 结果与讨论
2.1检测波长的选择
采用紫外可见光分光光度仪对一定浓度的香豆素及其衍生物溶液进行紫外波长全扫描(190~400 nm),紫外光谱图显示,在210 nm波长下,各物质的响应强度较高,但在色谱分析过程中发现,随着流动相中有机相比例的逐渐升高,基线漂移较为严重,因此未选择210 nm作为最佳吸收波长。参照各目标物质的紫外光谱图,选取的最优检测波长见“1.2”所述。
2.2流动相的优化
根据香豆素及其衍生物的化学结构及色谱保留行为相似的特点以及双香豆素弱酸性的性质,实验比较了甲醇-水、乙腈-水、乙腈-0.02 mol/L乙酸铵溶液(含0.2%乙酸)、乙腈-0.05 mol/L乙酸铵溶液(含0.2%乙酸)和乙腈-0.05 mol/L乙酸铵溶液(含0.25%乙酸)作为流动相时的分离效果。结果表明,香豆素类物质以甲醇-水洗脱时,随着有机相比例的提升,基线漂移较严重;以乙腈-水作为流动相时,基线较平稳,响应强度较甲醇-水高,但双香豆素的峰形前展严重;鉴于双香豆素呈弱酸性,在流动相中添加了不同浓度的乙酸-乙酸铵缓冲体系,结果表明,双香豆素在乙腈-0.05 mol/L乙酸铵溶液(含0.25%乙酸)流动相体系下的峰形较为对称,且各目标分析物实现了基线分离。香豆素及其衍生物的高效液相色谱图见图1。
2.3提取方法的优化
2.4质谱确证方法的优化
鉴于化妆品基质较为复杂的特点,在采用高效液相色谱法进行分析时,其中的基质成分可能会对目标物的测定造成干扰。因此,本研究对疑似阳性样品采用高效液相色谱-串联质谱法进行确认。实验结果表明,除双香豆素和醋硝香豆素在电喷雾负电离模式下获得较高丰度的[M-H]-前体离子外,其余6种目标物均在电喷雾正电离模式下有较高响应的[M+H]+前体离子。采用子离子扫描方式进行二级质谱分析,通过优化锥孔电压、碰撞能量等主要参数,均获得两个响应强度较高的产物离子。香豆素及其衍生物的质谱确证图谱见图2。
2.5方法的特异性
为考察本方法的特异性,对空白溶剂及25例经测定不含待测目标物的化妆水、膏霜、乳液、散粉和唇膏类样品进行测定,代表性色谱图见图3。在目标化合物出峰时间范围内,空白试样无基质干扰峰出现,表明方法建立的流动相洗脱程序合理,可有效避免样品分析时的基质干扰作用,提高方法对化妆品中香豆素及其衍生物检测的特异性。
2.6线性关系、检出限及定量下限
在最佳实验条件下,移取适量混合标准储备液,用乙腈逐级稀释成浓度分别为0.2,0.5,1.0,2.0,5.0,10.0,20.0,50.0 μg/mL的混合标准工作溶液,以色谱峰面积(y)对质量浓度(x,μg/mL)进行线性回归,绘制标准曲线。结果表明,香豆素及其衍生物在0.2~50.0 μg/mL浓度范围内呈良好的线性关系,相关系数(r2)不小于0.999 6(表2)。
向空白化妆品中添加不同浓度的混合标准溶液,充分涡旋混匀,按照本方法进行处理和分析测定,分别以3倍信噪比(S/N=3)和10倍信噪比(S/N=10)对应的目标物浓度作为方法的检出限和定量下限。结果表明,对香豆素加标水平分别为4 mg/kg和8 mg/kg的样品进行预处理和分析后,对应的色谱峰信噪比分别约为3和10。以相同方式进行样品分析,确定了其它7种目标化合物的检出限和定量下限。8种化合物的检出限为4~10 mg/kg,定量下限为8~20 mg/kg,结果见表2。
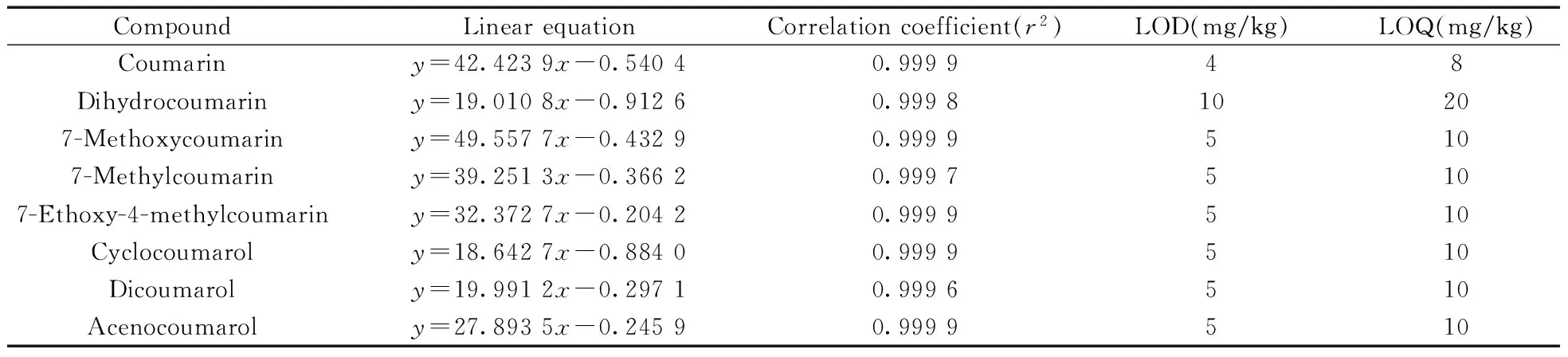
表2 香豆素及其衍生物的线性方程、检出限及定量下限
2.7稳定性考察
为考察目标分析物在本方法中的稳定性,分别制备0.5 μg/mL和5 μg/mL的基质匹配混合标准溶液,日内(0,3,9,12,15 h)伴随标准曲线进行测定,计算含量,得到日内相对标准偏差(RSD);连续测量5 d,计算得到日间RSD。经测定,香豆素及其衍生物的日内RSD及日间RSD均在0.5%~8.0%之间,表明方法具有较好稳定性。
2.8回收率与精密度
称取空白化妆品样品0.5 g(精确至0.001 g),分别添加低、中、高3个浓度的混合标准溶液,以伴随标准曲线进行定量,进行回收率实验,结果见表3。香豆素及其衍生物的平均回收率为84.6%~100.7%,RSD为0.8%~9.7%,表明本方法的准确度和精密度良好。
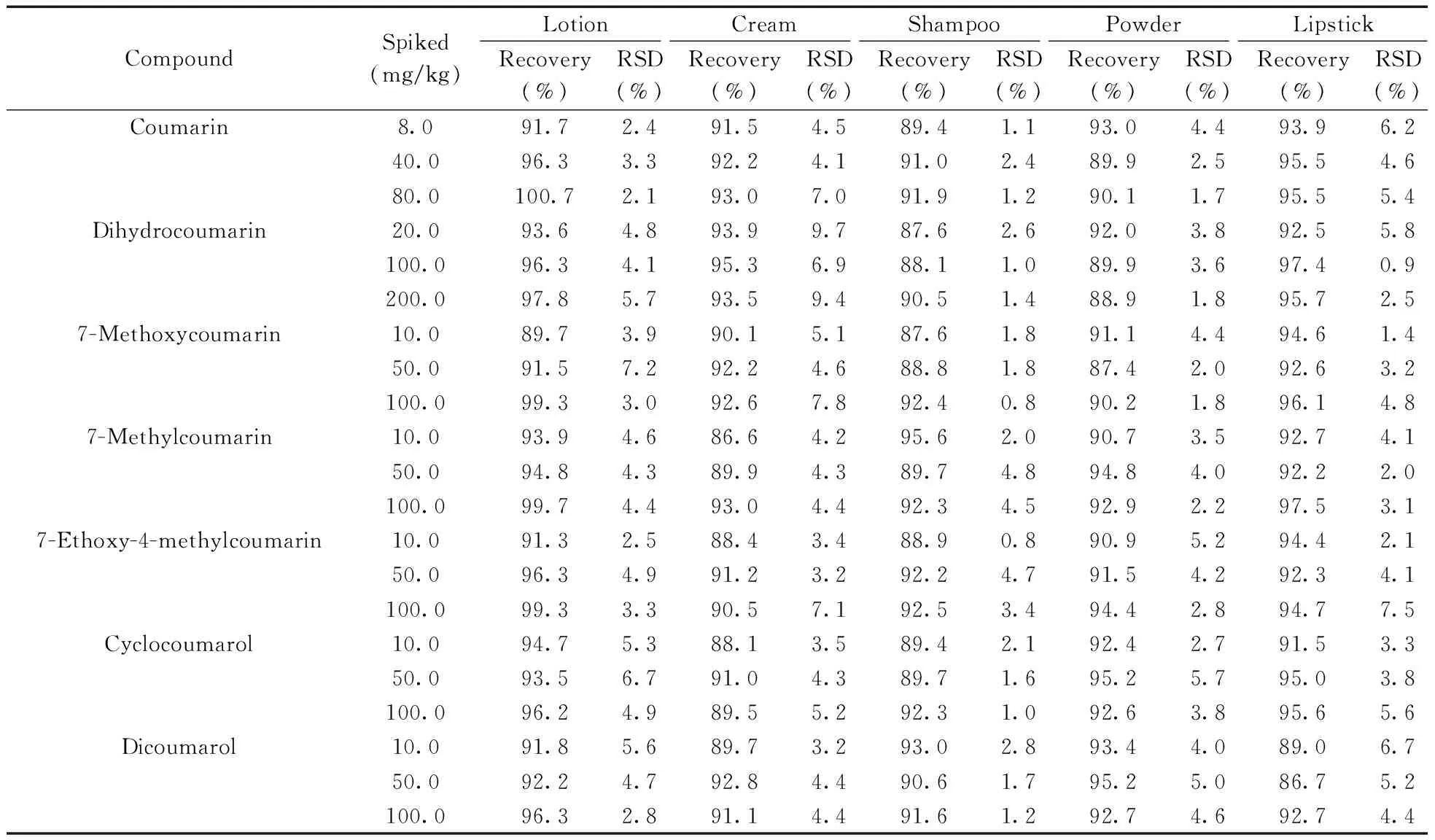
表3 化妆品中香豆素及其衍生物的加标回收率及相对标准偏差(n=6)
(续表3)
2.9实际样品测定
应用本实验建立的方法对市售的进出口化妆品(包括化妆水、膏霜、香波、散粉和唇膏类)共15例样品进行检测,均未检出目标分析物,并通过高效液相色谱-串联质谱法确证,无假阳性样品出现。
3 结 论
本研究建立了不同类型化妆品中致敏原香豆素及其衍生物的高效液相色谱测定方法,疑似结果以高效液相色谱-串联质谱进行确证,有效避免了假阳性结果。方法前处理简便、特异性强、灵敏度高且稳定性好,适用于化妆品中香豆素致敏原及其衍生物的测定。
[1]Liu J Y,Yang X D,Xu L Z,Yang S L.Chin.J.Chromatogr.(刘江云,杨学东,徐丽珍,杨世林.色谱),2002,20(3):245-248.
[2]Ma Q,Bai H,Wang C,Li W T,Ma H J,Li J R,Meng X S,Chen Y X.J.Instrum.Anal.(马强,白桦,王超,李文涛,马会娟,李晶瑞,孟宪双,陈云霞.分析测试学报),2014,33(3):248-255.
[3]Jiang J Q,Feng Y,Wang H M,Liu X Y,Zhang S W,Chen M Q.ActaPhysico-Chim.Sin.(江金强,冯艳,王红梅,刘晓亚,张胜文,陈明清.物理化学学报),2008,24(11):2089-2095.
[4]The Federal Institute of Risk Assessment(BfR)[2013-12-23].http://ww.bfr.bund.de/cd/template/index_en.
[5]Ehrenforth S,Schenk J F,Scharrer I.Semin.Thromb.Hemost.,1999,25(1):79-83.
[6]Directive 2003/15/EC of the European Parliament and of the Council of 27 February 2003 Amending Council Directive 76/768/EEC on the Approximation of the Laws of the Member States Relating to Cosmetic Products.http://eur-lex.europa.eu/Lex-UriServ/LexUriServ.douri=OJ:L:2003:066:0026:0035:en:PDF.
[7]Pan T L,Wang P W,Aljuffali I A,Leu Y L,Hung Y Y,Fang J Y.Toxicol.Lett.,2014,226(2):173-181.
[8]Ministry of Health of the People's Republic of China.Hygienic Standard for Cosmetics.http://www.gov.cn/zwgk/2007-01/26/content_508651.htm.
[9]Xi H W,Ma Q,Li Q,Lei H M,Bai H,Wang C.J.Instrum.Anal.(席海为,马强,李强,雷海民,白桦,王超.分析测试学报),2010,29(1):46-50.
[10]Xi H W,Ma Q,Wang C,Bai H ,Liu X,Wang Y.J.Instrum.Anal.(席海为,马强,王超,白桦,刘茜,王烨.分析测试学报),2010,29(12):1168-1172.
[11]Cheng Y,Wang C,Xue Y M,Chen W,Wang X,Bai H,Cai T P,Hu K X.J.Instrum.Anal.(程艳,王超,薛一梅,陈伟,王星,白桦,蔡天培,胡孔新.分析测试学报),2008,27(2):196-199,202.
[12]Yang R J,Wei B W,Gao H,Yu W J.Chin.J.Chromatogr.(杨荣静,卫碧文,高欢,于文佳.色谱),2012,30(2):160-164.
[13]Zhang W,Zhu Z J,Zhang H,Zhang Y X.Chin.J.Anal.Lab.(张薇,朱智甲,张晗,张宇霞.分析试验室),2012,31(5):104-108.
[14]Wisneski H H.J.AOACInt.,2001,84(3):689-692.
[15]Zhao X Y,Lin Y F,Hu X Z,Wang P,Fu X F,Li J.Chin.J.Anal.Lab.(赵晓亚,林雁飞,胡小钟,王鹏,付晓芳,李晶.分析试验室),2010,29(3):76-79.
[16]Li C Y,Li Z G,Zhou S Y,Ye D F,Liu W H.J.Chin.MassSpectrom.Soc.(李长于,李祖光,周示玉,叶丹凤,刘文涵.质谱学报),2011,32(5):265-270.
[17]del Nogal Sanchez M,Perez-Pavon J L,Moreno Cordero B.Anal.Bioanal.Chem.,2010,397(6):2579-2591.
Determination of Coumarin Allergen and Its Derivatives in Cosmetics by HPLC and Their Verification by HPLC-MS/MS
MENG Xian-shuang1,MA Qiang1*,YUAN Han-cheng2,BAI Hua1,ZHANG Qing1,GUO Xiang-yu1
(1.Chinese Academy of Inspection and Quarantine,Beijing100176,China;2.Waters Technologies(Shanghai),Ltd.,Shanghai201206,China)
A high performance liquid chromatographic(HPLC) method was developed for the determination of coumarin allergen and its seven derivatives,including dihydrocoumarin,7-methoxycoumarin,7-methylcoumarin,7-ethoxy-4-methylcoumarin,cyclocoumarol,dicoumarol and acenocoumarol in cosmetics.Cosmetic samples were ultrasonically extracted with 0.1 mol/L aqueous sodium hydroxide-acetonitrile(1∶9).The extract was centrifuged,and filtered by microporous membrane.The chromatographic separation for the analytes was achieved on a ZORBAX SB-C18(250 mm×4.6 mm,5 μm) column.Gradient elution was performed using 0.05 mol/L aqueous ammonium acetate solution(containing 0.25% acetic acid) and acetonitrile.The quantification was carried out by the external standard method,and the suspected samples were further verified by high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS).The experimental results indicated that good linearities were observed over a certain concentration range with correlation coefficients better than 0.999 6,and the limits of quantitation(LOQs) were between 8 and 20 mg/kg.The average recoveries for the analytes at three spiked levels ranged from 84.6% to 100.7% with relative standard deviations(RSDs) of 0.8%-9.7%.The proposed method is accurate,robust and specific,and could provide scientific basis and a technical support for the inspection and quality control of cosmetics.
coumarin and its derivatives;allergen;high performance liquid chromatography(HPLC);high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS);cosmetics
2015-12-19;
2016-02-01
国家科技支撑计划项目(2013BAK04B03);质检公益性行业科研专项项目(2012104013-4);国家标准化管理委员会计划项目(20130980-T-607)
马强,博士,副研究员,研究方向:消费品中化学危害物质检测技术及风险评估,Tel:010-53897463,E-mail:maqiang@caiq.gov.cn
doi:10.3969/j.issn.1004-4957.2016.07.004
O657.63;TQ658
A
1004-4957(2016)07-0799-06
