闪式真空热解(FVP)在有机合成中的应用
2016-08-16徐飘扬张前炎谢素原
徐飘扬,张前炎,谢素原,高 飞*
(1.闽南师范大学 化学与环境学院,福建 漳州 361000; 2.厦门大学 化学化工学院,福建 厦门 361005)
闪式真空热解(FVP)在有机合成中的应用
徐飘扬1,张前炎2,谢素原2,高飞1*
(1.闽南师范大学 化学与环境学院,福建 漳州 361000;2.厦门大学 化学化工学院,福建 厦门 361005)
闪式真空热裂解(Flash vacuum pyrolysis,FVP)是一种反应底物在真空条件下蒸发或者升华后迅速通过较高温度的热管道发生热解反应的过程. 该热裂解方法经常被运用于合成一些重要的非平面型芳香化合物,比如著名的心环烯C20H10(Corannulene),富勒烯C60等. 主要针对FVP方法的发展历史、装置的基本构成、反应的基本历程以及该方法在有机合成中的实际应用等方面进行了系统的综述. 相对于传统有机合成化学方法,FVP方法的优势在于可以提供更高的外界能量来帮助产物化学键的形成和更快速的冷却方式来帮助稳定反应所得到的产物,因此该方法不仅能高效、方便地合成得到一些常规有机合成方法不能轻易获得的目标化合物,还可以获得一些热力学极其不稳定的产物. 当然,FVP方法也有其限制,比如对于一些在真空条件下难以挥发的化合物FVP方法就不适用了,另外,因为所有FVP反应都是在气相条件下完成,所以该方法主要适用于分子内的消除或环合反应,对于有机合成中普遍存在的双分子反应以及多分子反应也难以通过FVP方法来实现,但作为一类独特、实用的有机合成方法,FVP在有机合成中得到了较广的应用和不断地发展.
闪式真空热解;有机合成;气相合成;分子内反应
早在19世纪,有机化学家们就开始尝试将有机化合物蒸馏或者升华通过炙热的管道进行热解. 随后,HURD发表了经典专著“ThePyrolysisofCarbonCompounds”[1],这本经典著作总结了当时所有的热解过程,为当时的有机化学提供了许多有价值的信息并产生了深远的影响. 随着石油石化工业的兴起,各种大规模高温流动裂解反应在工厂中得到不断应用,而实验室的热解反应仅仅限制在少数几个挥发性液体化合物的热解. 20世纪中期,随着气相反应和气体动力学的迅速发展,烷基自由基的重要性被逐渐认识. 到了1980年左右,科研工作者通过电子轰击质谱分析分子的碎片,发现了一些新奇和意想不到的活性中间体,这些活性中间体对认识不同化学反应的机理历程方面具有非常重要的意义,因此化学家们开始热衷于对化学反应的活性中间体的探究,而低压热解(very low pressure pyrolysis, VLPP)方法恰好能够在非常短的时间内通过高温反应区生成活性中间体[2],于是化学家们又开始把目光重新转向热管加热的方法. 为了尽量达到和电子轰击类似的高能量,快速热裂解反应要求在高温低压的条件下发生,这种方法后来被称为闪式真空热解方法(Flash Vacuum pyrolysis, FVP).
由于FVP方法是一种将反应物在真空条件下蒸发或者升华后迅速通过较高温度的热管道而发生热裂解,因此整个热裂解反应都是在气相中完成,和普通的气相反应比较,FVP采用了高真空度的环境,使得气态的反应底物分子十分迅速地通过高温区域,因此底物分子在高温区域的时间非常短. FVP其中的一个关键技术就是使用惰性气体当载气. 惰性气体一般不仅自身在灼热石英管中不会发生反应,而且惰性气体可以包裹保护底物分子,使其能更好地发生分子内反应. 由于底物分子在高温区域的接触时间相对于其他热解方法是非常短的,因此一些热力学不稳定的活性反应组分可以在随后的冷阱中得到稳定并被收集用于后期分离表征[3-5].
实验室中大部分的有机化学反应是在液相中进行,也就是说反应物通常需要溶解在有机溶剂中发生相互反应. 因为溶剂沸点的原因,其反应的温度受到很大制约,通常在干冰的升华点-78 ℃到一些高沸点溶剂沸点(300 ℃左右)之间. 对于一些需要高能量的反应,液相反应的劣势比较明显. 相比于有机合成中的液相反应,FVP条件所提供的温度是远远大于传统有机化学反应. 根据反应所需能量,FVP装置的温度最高可以提高至1 250 ℃. FVP反应中的“温和”温度在350~650 ℃范围内,在这种温度条件下,化合物的大多数官能团仍然能较好的保存下来,并没有发生任何转变,其中有些不含活泼官能团的芳香化合物在温度超过1 000 ℃时仍然很稳定.
另外,对于一些温度达不到太高的有机液相反应,一般是通过延长加热时间的做法来尝试能否获得目标产物. 但是加热时间的延长不一定利于反应,反应物由于长时间处于反应体系当中,这就有可能使得反应物与反应物之间、反应物与产物、产物与产物之间,甚至和溶剂分子之间也会发生复杂的分子间反应. 同时,又因为持续加热的缘故,生成的产物有可能发生二次反应. 由于液相反应温度的限制使环境提供不了反应所需的能量来越过能垒,延长时间也会发生二次反应. 为了避免二次反应的发生,在有机化学合成中有时使用一些催化剂来改变反应机理降低反应所需要的能量,但是催化剂对于降低反应能垒是有限度的. 尽管找到了合适的催化剂,其制备、保存也会有问题,还有某些催化剂的使用条件受到严格的限制(比如溶剂,pH值,无水无氧等). 相比较而言,FVP的反应过程可以理解成气相单分子自我重组的分子内反应,完全不受溶剂,催化剂的影响,也不用担心复杂的分子间反应、二次反应等带来副反应的发生.
通过以上比较得出,FVP的微观过程就是反应底物分子蒸发或者升华然后被载气运输通过高温区而发生热裂解或重排反应. 由于底物分子被气化而且真空度较高,理论上就可以认为反应物分子是一个一个地通过高温区,分子瞬间受热使得分子强烈振动,通常发生分子内反应生成产物,随后被液氮迅速冷却. FVP体系相对于普通的热解体系需要更高的温度,反应底物通过激烈短暂的热激发后产物立即被冷却,由于体系中的高真空和惰性气体保护,该方法可以简单高效地获得常规有机反应难以合成的产物. 本文主要针对FVP方法的发展历史、FVP装置的基本构成、FVP反应的历程、FVP方法在有机合成中的实际应用等方面进行系统的综述.
1 FVP实验装置
FVP反应实验的基本装置如图所示,是简单而容易实现的[6]. 在真空泵的作用下整个体系达到高真空的状态,反应基质蒸发通过被管式炉加热至固定温度的炉管进行反应. 通过了炉管末端的产物急剧冷却附着在内壁上. 待反应结束后,可以通过溶剂清洗内壁获得产物.
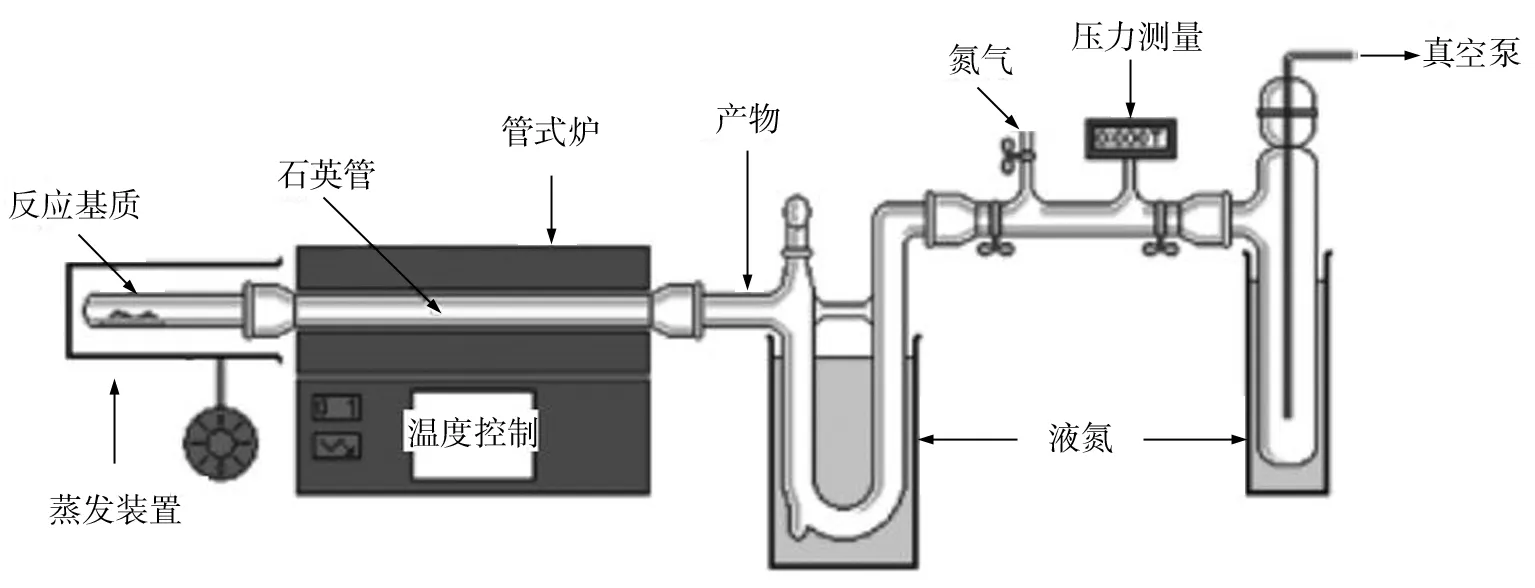
图1 FVP反应装置Fig.1 FVP apparatus
如果反应管是耐热玻璃管,在温度达到520 ℃左右就会开始软化,这将大大限制FVP的应用,如果替代成石英管,高温区反应温度最高可以达到1 250 ℃,这样就可以充分发挥FVP反应的优势. 为了延长反应物通过高温区的接触时间,炉管的中间有时也可以填充一些惰性材料(石英棉等),此时通常稍微地降低高温区的温度会达到更理想的效果. 此外,还可借助填充催化材料来实现FVP高温催化反应,但由于是气固两相反应,目前这方面研究的比较少. 另外在反应的压力方面,FVP反应实验需要一个相对稳定的真空环境,一般来说高强度的回转泵(0.1~1.0 Pa)就可以满足. 如果需要研究反应中间产物或者活性中间体的话,就可能需要更低的真空度.
综上所述,FVP反应的主要参数有三个,底物的挥发温度(控制物料通过速度),管式炉的温度和背景真空度. 实际上,泵的输出压力一般比较稳定,物料通过速度对FVP反应的影响也不是很大. 在这种情况下,管式炉的温度成为唯一的变量. 而底物的转化率和炉温的关系通常呈现一个有规律的S型曲线.
2 FVP反应历程
从反应历程上分析,由于离子电离能比较高,又没有溶剂作用,所以在FVP反应条件下不存在离子中间体,大多数FVP反应是周环反应(电环化,逆Diels-Alder反应)、自由基反应或者双自由基反应(卡宾、氮烯、苯炔). 这些过程的典型例子如图2~5所示. 比如,图2显示的FVP反应属于周环反应的电环化过程,该过程被认为是通过脱离一些稳定的小分子(例如HCl,N2,CO2等)来重排分子[7]形成芳香化合物.
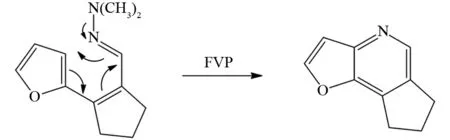
图2 FVP反应中的电环化反应Fig.2 Electrocyclizationreaction in FVP reaction
又如图3所示,两个C-C单键的断裂被认为是经过逆Diels-Alder协同发生,一步反应得到产物[8].
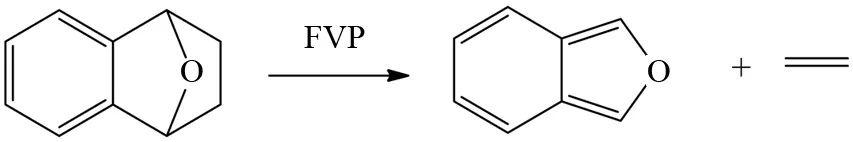
图3 FVP反应中的逆Diels-Alder反应Fig.3 Retro-Diels-Alder reaction in FVP reaction
虽然FVP方法反应底物分子的相对浓度是非常稀的,分子内反应通常占主导地位,但是一些非常活泼的中间体之间还是有发生分子间反应的可能,比如图4所示的苯炔中间体的分子间自我偶联反应[9].
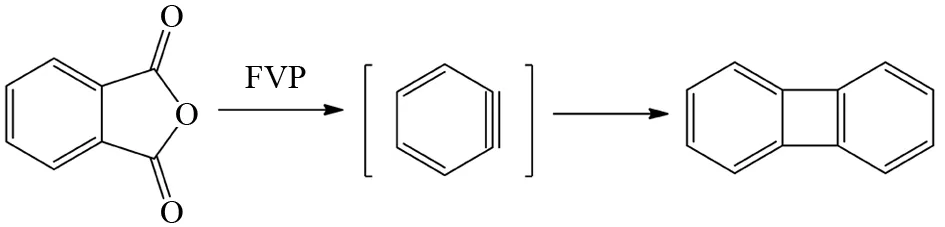
图4 FVP反应中的双分子偶联反应Fig.4 Bimolecular coupling reaction in FVP reaction
另外,自由基或分子内双自由基的重排也是FVP中一类典型反应[10](图5).
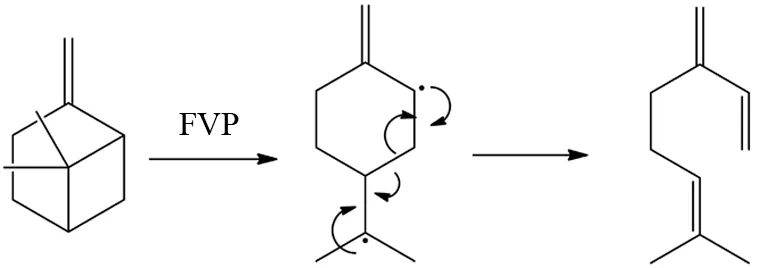
图5 FVP反应中的双自由基重排反应Fig.5 Biradical rearrangement in FVP reaction
FVP实验和电子轰击质谱实验都是瞬间提供高能量给化合物分子,但是这两种实验方法对于相同的化合物分子有时候会得到完全不同的结果. 一般来说,FVP反应是从分子中作用力最弱的化学键开始断裂,而电子轰击质谱实验是由形成正离子自由基稳定性所决定. 通过图6就能看出两者的差别[11].
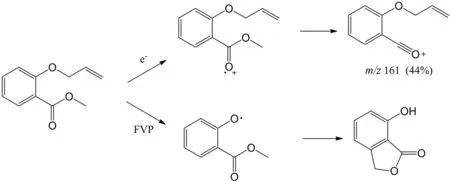
图6 FVP反应与电子轰击质谱的差别Fig.6 Difference between FVP reaction and electron impact mass spectra
3 FVP在有机合成化学中的应用
3.1烷烃
由于C-C单键在高温时容易发生断裂,FVP反应很少应用于只产生C-C单键的反应,所以这类FVP反应通常在相对比较低的温度下才能实现. 这类反应拥有共同的特点:通常反应底物分子存在比较容易消除的原子或小分子基团. 倘若提供合适的能量,小分子基团会从底物分子中离去发生分子内消除反应,然后底物分子重新组合形成新的C-C单键.
比如,TRAHANOVSKY等发现FVP热解草酸酯底物分子时,该分子会先脱去2个二氧化碳分子生成苄自由基,然后经过分子间耦合就可以生成联苄及其衍生物[12](图7).
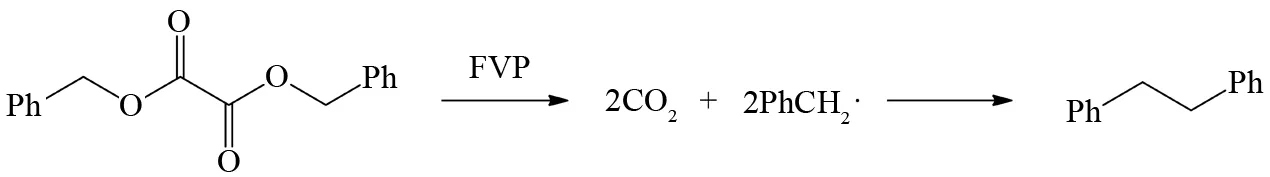
图7 草酸脂衍生物的FVP反应Fig.7 FVP reaction of oxalate ester derivatives
同样,LEONARD等也发现苄基砜类底物在FVP条件下发生分子内热解脱去SO2分子后形成苄基自由基,然后经过分子内自由基耦合得到目标产物,该反应已经广泛用于不含任何杂原子的环烷的合成[13](图8).
相似地,BROWN等发现苄基酮类反应底物在FVP条件下先脱去CO分子也可以产生苄自由基,然后由分子内自由基耦合形成C-C单键,从而形成多桥芳烃[14](图9). 从图8和9可以看出,其他常规有机合成手段难以实现的多桥芳烃化合物,但通过FVP反应产生苄自由基的策略可以方便地合成得到多桥芳烃化合物[15-16].
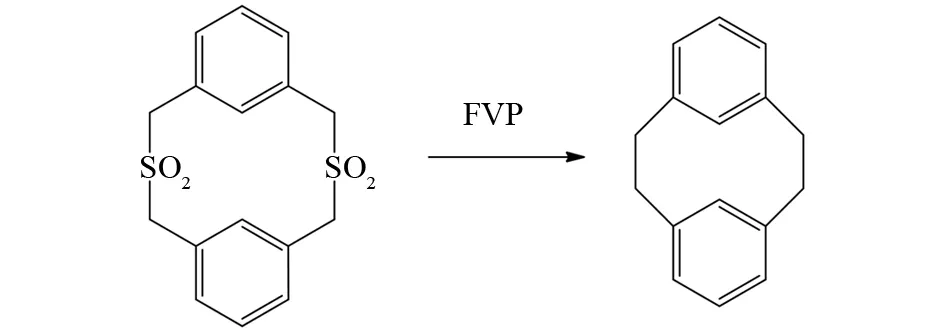
图8 苄基砜类化合物的FVP反应Fig.8 FVP reaction of benzyl sulfone compounds
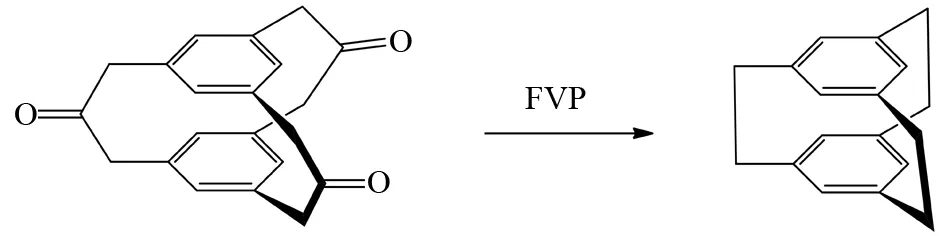
图9 苄基酮类化合物的FVP反应Fig.9 FVP reaction of benzyl ketone compounds
图8和9中显示,其反应底物所脱去的小分子在底物分子骨架中都是相连的,而BOEKELHEIDE等研究发现未连接的小分子也可以在分子内共同离去. 比如把1-氯甲基-2-甲基苯作为FVP反应底物,显然该底物分子的H原子、Cl原子并没有直接相连,但它可以通过逆电环化的形式离去HCl分子生成双烯中间体,再由双烯中间体经过电环化生成苯并环丁烯[17](图10),该反应达到了85%产率. 另外RUEDI等发现2-甲基苯乙酰氯在FVP条件下也经过类似的反应机理得到苯并环丁酮,其产率高达到80%[18](图11).
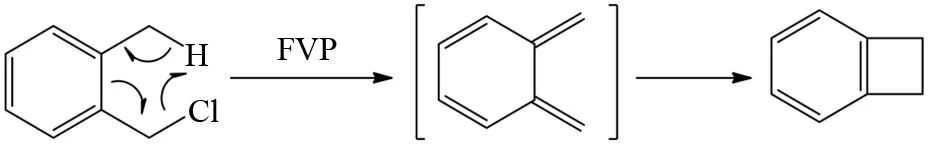
图10 1-氯甲基-2-甲基苯的FVP反应Fig.10 FVP reaction of 1-(chloromethyl)-2-methylbenzene
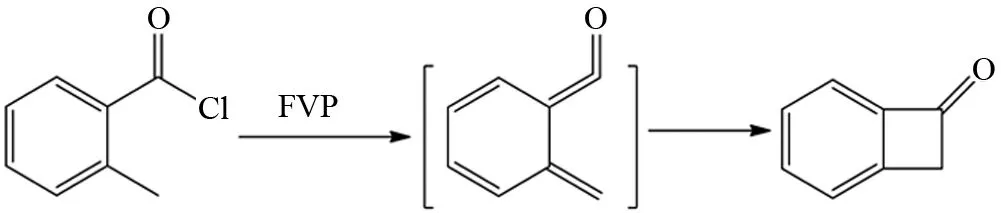
图11 2-甲基苯甲酰氯的FVP反应Fig.11 FVP reaction of 2-methylbenzoyl chloride
3.2烯烃
由于碳碳双键的键能比其他类型的单键(例如碳碳单键、碳氧单键等)的键能高,所以碳碳双键在高温下的稳定性比单键更好,因此FVP方法经常被用来合成一些含有双键的化合物. 在FVP反应条件下,烯烃通常由以下几种方式制备得到:1)几个单键之间相互作用发生逆Diels-Alder或逆电环化等协同反应[19],2)从键能相对较低的单键开始断裂引发整个分子骨架进行重排反应,3)分子内消除易离去的小分子形成新的烯烃分子.
比如,乙烯和丁二烯发生反应产生环己烯是一个经典的Diels-Alder反应,而环己烯在FVP条件下可以发生逆Diels-Alder反应生成乙烯和丁二烯产物(图12).
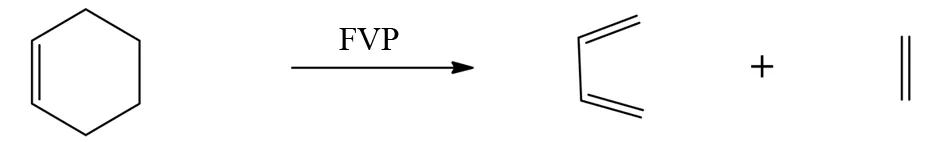
图12 环己烯的FVP反应Fig.12 FVP reaction of cyclohexene
JENNESKENS等发现了一系列乙酸酯类底物分子在FVP条件下会通过逆电环化反应机理生成烯烃和羧酸(图13). 而黄原酸酯(X=O,Y=S,Z=SCH3)类底物分子在FVP条件下也可以发生相似的反应,而它的热解温度要低于相应的乙酸酯(X=Y=O,Z=CH3)[20].
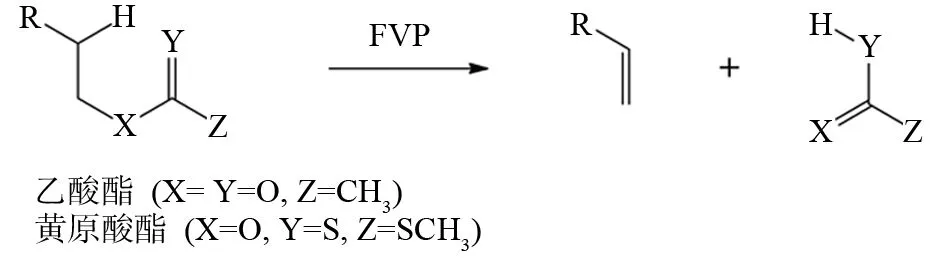
图13 乙酸酯类和黄原酸酯化合物的FVP反应Fig.13 FVP reaction of acetic acid ester and xanthate compounds
乙酸酯类底物分子在FVP条件下生成烯烃的优势在于可以制备一些热力学不稳定的烯烃,因为这些不稳定的烯烃产物可以在FVP的低温收集冷阱里得到冷却保护,可以避免不稳定的烯烃发生自身聚合或分解反应. 而在普通的有机合成反应中,这些生成的不稳定烯烃会因为进行二次副反应难以成功制备得到. 例如,不稳定的二氢噻吩化合物在传统有机合成条件下是难以获得的,TRAHANOVSKY等通过FVP反应制备二氢噻吩的产率可以达到85%[21](图14).

图14 不稳定的二氢噻吩化合物的FVP反应Fig.14 FVP reaction of the unstable dihydrothiophene compounds
值得一提的是,FVP反应不仅可以获得热力学不稳定烯烃分子,FVP反应还可以制备具有立体选择性的烯烃[22]. 如图15、16所示,呋喃或环戊二烯加合物三环骨架在FVP反应中发生逆Diels-Alder反应,生成的产物分子中的两个手性碳原子的手性都未发生手性变化,保持了原有的立体选择性[23]. 通常三环骨架呋喃加合物(X=O)在FVP条件下发生逆Diels-Alder反应所需的温度低于环戊二烯加合物(X=CH2).
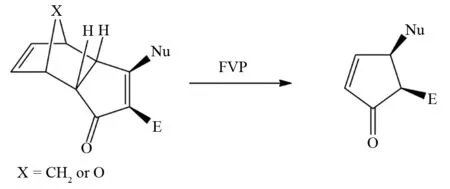
图15 三环骨架在FVP反应中的应用Fig.15 Application of three ring skeleton in FVP reaction
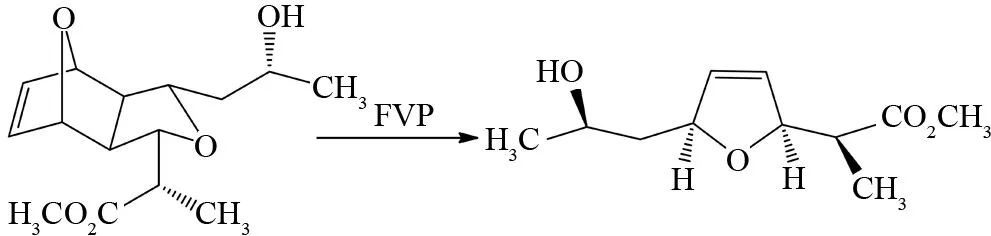
图16 三环骨架呋喃加合物的FVP反应Fig.16 FVP reaction of furan adduct bearing the tricyclic skeleton
除了单烯烃可以通过FVP方法合成之外,双烯以及多烯烃也可以利用FVP手段合成得到. 如MEHTA等发现了笼状结构的双酮分子,通过断裂两个桥键,然后空间重排获得双烯双酮分子[24](图17). BRAIN等则发现多烯烃可以通过底物分子内脱去一个稳定小分子后形成,如图18所示,亚砜类底物通过离去一个一氧化硫分子,得到的产物比底物分子多出两个碳碳双键[25].
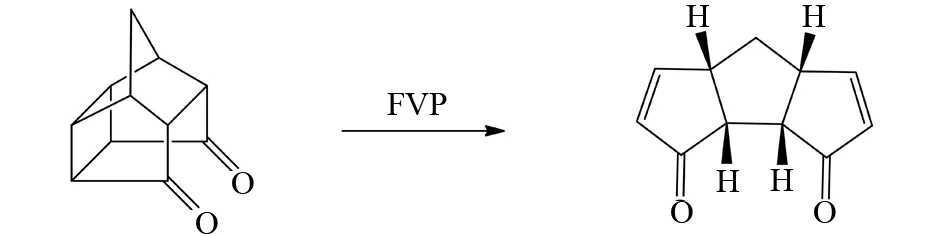
图17 笼状结构双酮分子的FVP反应Fig.17 FVP reaction of the cage structure with double ketone molecule
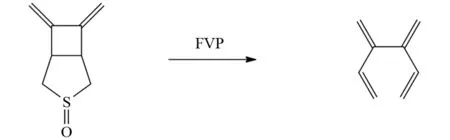
图18 亚砜类化合物的FVP反应Fig.18 FVP reaction of sulfoxide compounds
通过FVP产生多烯的另外一个重要过程是形成邻亚二甲基中间体,邻亚二甲基中间体是一个非常活泼的中间体,如同图10所示,邻亚二甲基中间体会发生电环化反应生成苯并环丁烯产物. 在杂环体系中,由于杂原子的孤对电子的影响,使得亚二甲基中间体具有稳定的共轭体系,因此杂环分子的双烯形式比环丁烯更稳定. 例如,TRAHANOVSKY等发现呋喃衍生物通过类似乙酸酯的逆电环化反应,经FVP反应生成呋喃亚二甲基,该中间体在-60 ℃时可以稳定存在在溶液中,在常温下通过头碰头的形式二聚得到多烯产物[26](图19).
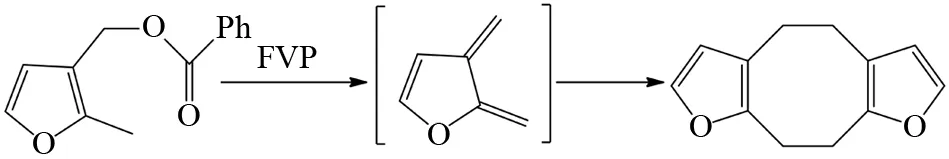
图19 呋喃衍生物的FVP反应Fig.19 FVP reaction of furan derivatives
SCHIESS等通过相似的方法,将1,3,5-三氯甲基-2,4,6-三甲基苯通过FVP反应离去三分子的HCl得到稳定的不饱和价态异构体[6]轴烯[27](图20).
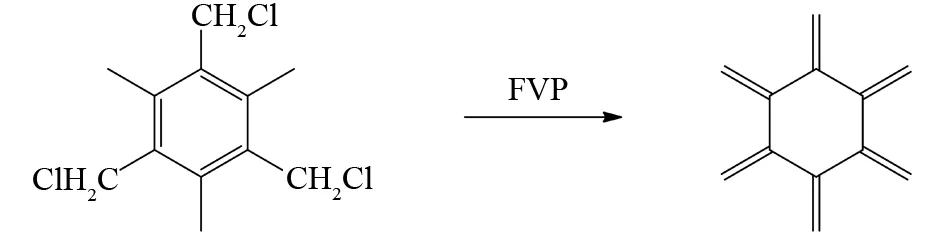
图20 1,3,5-三氯甲基-2,4,6-三甲基苯的FVP反应Fig.20 FVP reaction of1,3,5-tris(chloromethyl)-2,4,6-trimethylbenzene
累积多烯在常规有机合成中通常需要繁琐的反应步骤才能合成得到,FVP条件下,累积多烯可以简单地通过逆Diels-Alder方法获得. RIPOLL等将蒽加合物三环骨架通过FVP反应发生逆Diels-Alder反应,便可获得累积戊烯,该反应产率可以达到70%[28](图21). 累积多烯的性质比较活泼,但是FVP反应都是低温冷却下收集产品,所以生成的累积多烯可以避免发生二级副反应[29].
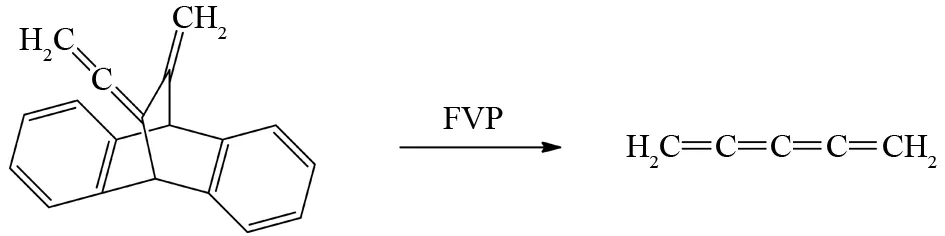
图21 蒽加合物三环骨架的FVP反应Fig.21 FVP reaction of anthracene adduct with the tricyclic skeleton
3.3炔烃
FVP反应制备炔烃的原料一般都是烯烃化合物,烯烃末端碳原子在FVP条件下先生成亚甲基卡宾中间体,然后碳碳双键上的氢原子发生1, 2-迁移至末端碳原子上产生炔烃,由于适合产生亚甲基卡宾中间体的底物不多,FVP反应用于合成炔烃的例子不算多. 如图22、23所示,麦氏酸(Meldrum’s acid)[30]和恶唑酮[31]的热解可以生成亚甲基卡宾中间体,然后亚甲基卡宾中间体通过分子内的重排得到炔烃,显然通过这种机理获得的炔烃都是末端炔烃. 由于末端炔烃在较高的FVP温度下,会可逆生成亚甲基卡宾,所以FVP条件下制备末端炔烃的反应温度也不能太高.
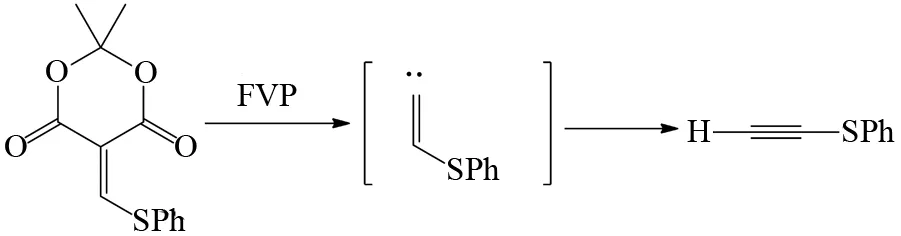
图22 麦氏酸的FVP反应Fig.22 FVP reaction of Meldrum’s acid
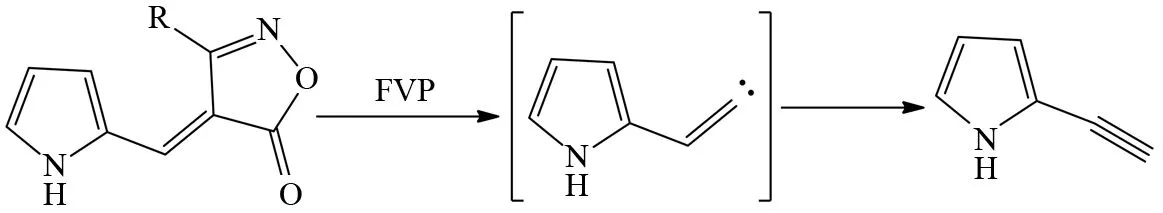
图23 恶唑酮的FVP反应Fig.23 FVP reaction of Oxazolone
相对于卡宾中间体只能生成末端炔烃化合物方法的局限性,AITKEN等通过邻羰基膦叶立德在500~750 ℃FVP反应中先发生电环化生成四元环中间体,然后再脱去三苯基氧膦就可以产生一系列非末端炔烃[32](图24).
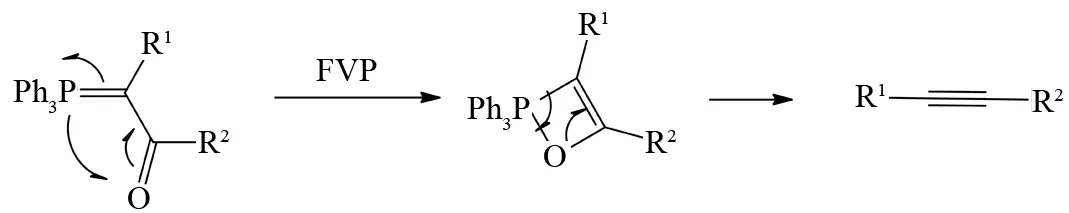
图24 邻羰基膦叶立德的FVP反应Fig.24 FVP reaction of ortho carbonylphosphonium ylide
3.4芳香族
如上所述,FVP方法可以用来制备烷烃、烯烃、炔烃化合物,但实际上FVP方法最成功之处是可以用于合成具有多种不同功能和结构的芳香族化合物. 例如,富勒烯C60的全合成、碗状碳氢化合物C20H10,C50H10的合成等等都是FVP方法的典型应用,尤其富勒烯C60的全合成的成功更是引起了科学家们对FVP方法的广泛关注[33]. FVP应用于合成芳香族化合物的例子举不胜数,本文仅就苯环型、非苯环型、碗状芳香族化合物中的某些成功例子重点讨论. 根据多环芳烃结构的不同,在FVP条件下经历的反应机理也会有不同,一般包括电环化、自由基环化、卡宾的插入或加成等.
例如,BROWN等将Meldrum酸衍生物通过FVP反应生成亚甲基烯酮重要中间体,亚甲基烯酮重要中间体先经过分子内的两步电环化反应产生酮,然后经过烯醇互变异构得到了多一个环结构的β-萘酚衍生物[34](图25). 通常萘的β位反应活性比α位的选择性差,所以β位取代的萘衍生物难以合成,因此该方法为β位多环酚等衍生物的合成提供了一个高效的合成方法[35].
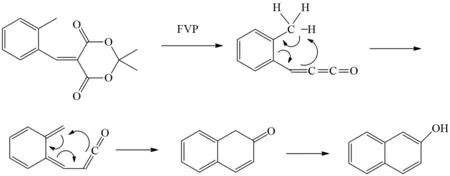
图25 Meldrum酸衍生物的FVP反应Fig.25 FVP reaction of Meldrum’s acid derivative
1-萘酚衍生物的FVP反应同样经历了上述反应机理. 8-甲基-1-萘酚由烯醇互变异构和逆电环化重排生成一个乙烯酮中间体,再由这个重要的乙烯酮中间体逆电环化重排和烯醇互变异构生成6-甲基-1-萘酚,这个反应过程揭示了8-甲基-1-萘酚和6-甲基-1-萘酚之间存在一个热力学平衡[36](图26).
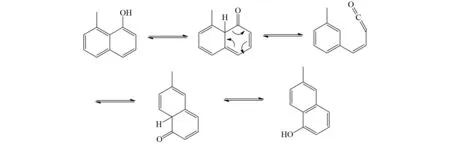
图26 8-甲基-1-萘酚和6-甲基-1-萘酚的互变异构Fig.26 Tautomerizm between 8-methylnaphthalen-1-ol and 6-methylnaphthalen-1-ol
在高温下,五元并七元碳环单元特别容易发生分子内重排反应. 对这类高温过程的分子内反应机制的探索是非常重要的,有利于理解富勒烯和碳纳米管的形成机理. 例如,甘菊环[37]在FVP条件下通过分子内部的重组得到性质更稳定的萘分子(图27).
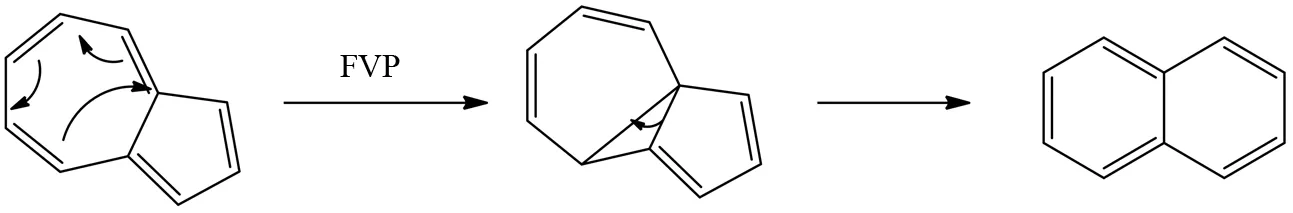
图27 甘菊环的FVP反应Fig.27 FVP reaction of azulene
多环芳烃底物分子内如果存在一些容易离去的小分子,在FVP条件下,这些小分子(如N2等)会离去产生芳烃自由基,然后芳烃自由基耦合,可以形成一系列结构有趣的多环芳烃. 例如苯并[c]噌啉在FVP反应条件下,先脱去一个N2分子,然后分子内自由基耦合形成四元环反芳香性多环芳烃[38](图28).
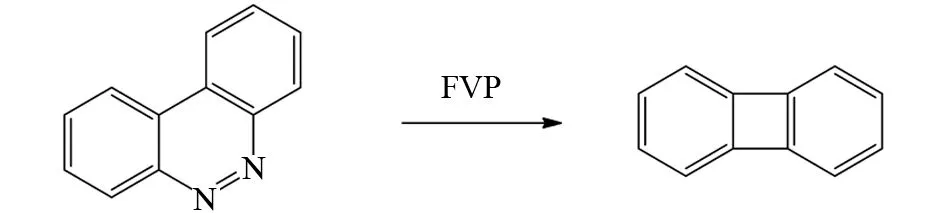
图28 苯并[c]噌啉的FVP反应Fig.28 FVP reaction of benzo[c]cinnoline
相对于反芳香性化合物,FVP反应还可以制备一些不稳定的“非苯型的”芳香族化合物. 叠氮化苄在FVP条件下先脱去氮气分子生成苄基卡宾,然后苄基卡宾通过分子内的插入反应形成环庚三烯自由基,最后两分子的七元环自由基之间相互耦合得到“非苯型的”二聚多环芳烃[39](图29).
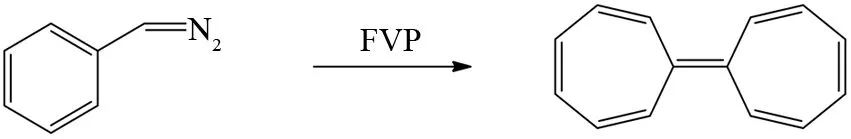
图29 叠氮化苄的FVP反应Fig.29 FVP reaction of diazomethylbenzene
多环芳烃底物中末端炔烃部位在FVP条件下也可以发生1,2-H迁移得到亚甲基卡宾. 例如,BROWN将2-乙炔基联苯通过FVP反应在炔基末端生成亚甲基卡宾,亚甲基卡宾和另一个苯环发生插入反应,以78%的产率获得主产物菲,另外通过卡宾和邻位苯环发生加成然后电环化作用可获得副产物苯并[a]甘菊环,其产率达到22%(图30).
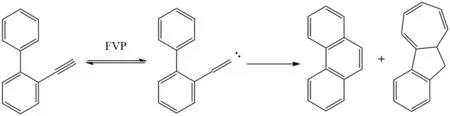
图30 2-乙炔基联苯的FVP反应Fig.30 FVP reaction of 2-ethynyl-1,1′-biphenyl
在FVP方法合成碗状多环芳烃方面,美国科学家SCOTT课题组做了许多令人印象深刻的研究工作. 其中具有代表性的是碗状碳氢化合物C20H10,C50H10和富勒烯C60的全合成. 这些工作共同的特点是通过FVP装置快速、高温加热一些设计合理的多环芳烃前体分子. 前体分子通过断裂C-H键或C-X(X=Cl,Br)键形成性质活泼的芳基自由基或卡宾,然后芳基自由基发生分子内环化或卡宾发生插入和加成反应.
在FVP反应例子中,芳香卤素化合物为合成碗状碳氢化合物和富勒烯碎片提供自由基来源的方法是非常高效的[40],因为碳卤键的键能比碳氢的键能小,所以热解脱卤化氢过程通常比未被取代的碳氢化合物的脱氢效率更有效,而且卤素原子还能起到定位环化的作用,因此,分子内新的C-C键可以通过碳卤键均裂生成的芳基自由基发生环化的过程形成. 如图31所示,完全由碳原子氢原子组成的多环芳烃通过FVP生成C36H12的产率只有0.6%,而此多环芳烃底物经氯化修饰后,在FVP反应条件下,产率可以达到25%[41].
SCOTT等发现卤素虽然能起到定位环化作用[42],但是卤素分子不一定非要在定位点碳原子上,可以在所需位点的邻位碳原子上. 如图32所示,底物分子先断裂C-Br键生成芳烃自由基,再经过芳基自由基之中的1,2-氢转移,使得自由基到达邻位碳上,然后与另一个芳环发生耦合. 所以原料中卤素的定位环化作用要考虑分子整体的空间结构[43].
SCOTT课题组将含有2个末端炔多环芳烃底物通过1 000 ℃高温的FVP反应先产生两个亚甲基卡宾,然后卡宾插入苯环成环,最后生成目标产物心环烯. 由于亚甲基卡宾有一部分通过卡宾对邻芳环的加成后发生电环化生成副产物,所以只能获得10%产率的心环烯[44]. 后来使用氯代烯取代末端炔官能团,氯代烯官能团在FVP过程中均裂碳氯键形成烯自由基,通过氢原子的1,2-迁移,产生末端烯自由基,最后通过和苯环的脱氢稠合作用形成心轮烯. 因此可以把产率提高到35%~40%[45](图33).
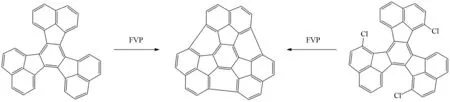
图31 C36H12的FVP合成Fig.31 Synthesis of C36H12by FVP reaction
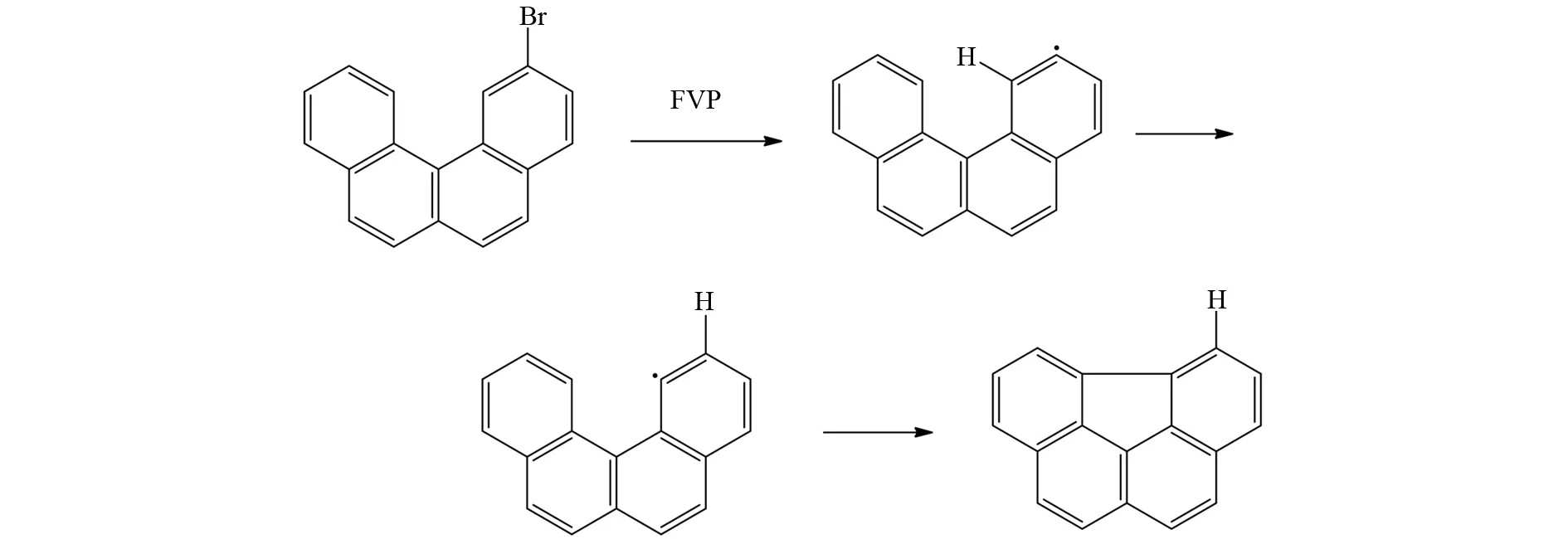
图32 FVP反应中的1,2-氢转移Fig.32 1,2-Hydrogen shift in FVP reaction
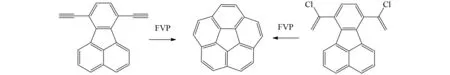
图33 心环烯Fig.33 Corannulene
IMAMURA等利用类似的过程,合成了硫杂碗型的分子[46]. 将底物分子通过FVP反应首先脱去氯自由基形成烯自由基,再通过烯上氢原子的1,2-迁移作用形成末端烯自由基,最后末端烯自由基通过和噻吩脱氢稠合形成目标产物(图34).
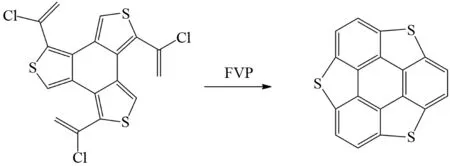
图34 硫杂碗型分子Fig.34 Trithiasumanene
SCOTT课题组在心轮烯的边缘进一步修饰上5个具有C5对称性的2,6-二氯苯基官能团,其在FVP反应条件下会优先断裂碳氯键得到芳基自由基,芳基自由基接着和心轮烯上的氢发生脱氢稠合形成5个新五元环,从而拉近了外围5个苯环的距离,最后外围的5个苯环自由基通过脱氢稠合形成碗状化合物C50H10[47](图35).
SCOTT等通过设计C60的前体分子C60H27Cl3,同样利用芳烃自由基环化机理,在FVP反应条件下首次完成了富勒烯C60的全合成. 该前体分子在FVP条件下脱去卤素生成自由基,然后通过类似的1,2-氢转移得到新的芳基自由基,新的芳基自由基接下来通过相互耦合以及在连续引发的自由基作用下,先耦合成碗状分子后逐步形成笼状得到最终产物C60[33],产率为0.1~1%(图36).
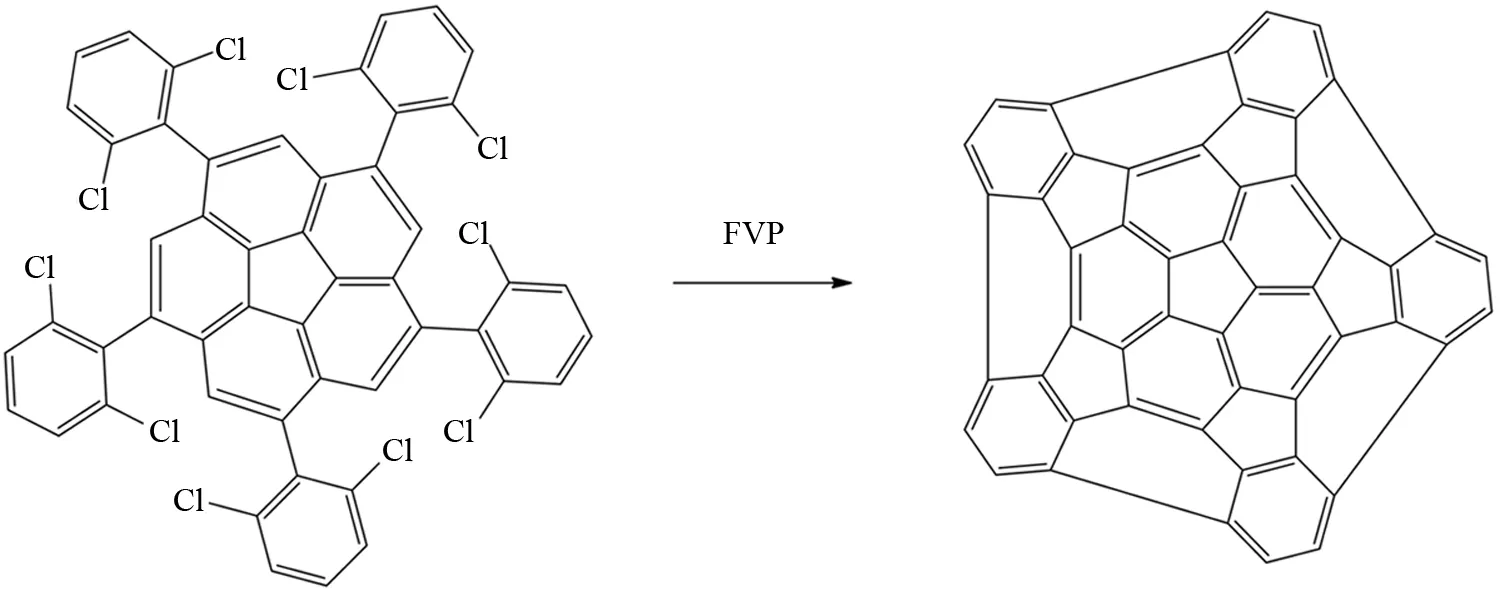
图35 通过FVP反应合成碗状化合物C50H10Fig.35 Synthesis of C50H10 by FVP reaction
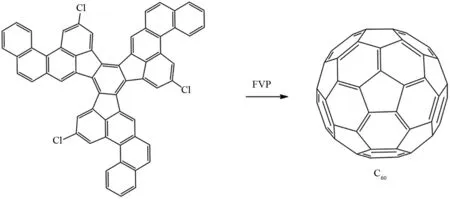
图36 通过FVP反应合成富勒烯C60Fig.36 Synthesis of fullerene C60 by FVP reaction
此后MARTIN等借鉴SCOTT等的方法设计并合成了富勒烯C78的前体分子C78H48[48]和C84的前体分子C84H42[49](图37),然后将它们通过FVP反应装置,所得的产物通过质谱检测到其目标分子的质谱信号,但由于目标产物的含量太少,在经过高效液相色谱分离后并未获得宏观量的目标产物C78和C84.
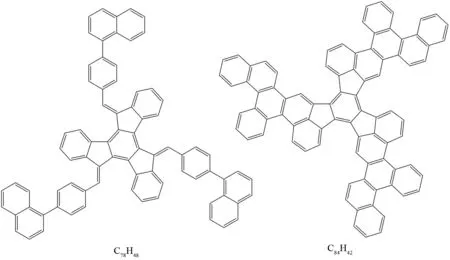
图37 C78H48和C84H42的结构Fig.37 Structure of C78H48 and C84H42
3.5杂环化合物
在FVP条件下,四元、五元、六元杂环化合物都可以生成[50-51]. 其中,四元杂环化合物由于存在较大的张力导致其非常不稳定,因此,在常规有机反应条件下难以合成,但FVP可以作为一种有效的方法产生四元杂环化合物. 由于FVP拥有反应的时间非常短,液氮冷却下收集产品等优势. 性质活泼的四元杂环化合物得以在这种相对极端的条件下稳定存在. 生成四元杂环化合物的机理一般都是分子内脱去一些易离去的小分子.
例如FVP早期应用的一个经典例子,ADGER等利用底物分子苯并三唑在FVP条件下高温分子内脱去一分子N2形成苄腈自由基,然后形成四元氮杂环产物. 该产物化学性质相当活泼,得在-78 ℃条件下完成收集、表征(图38).
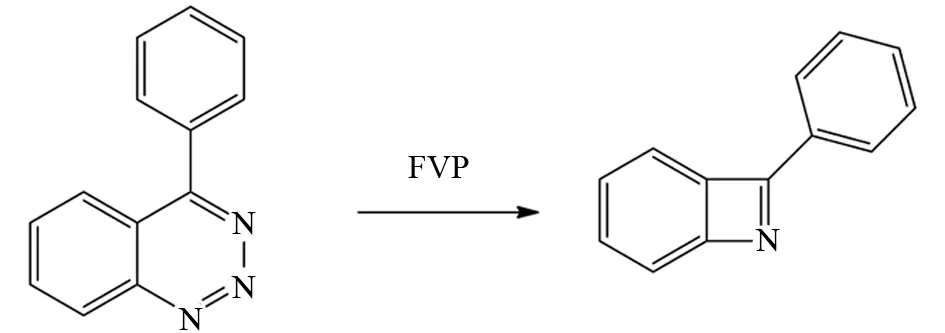
图38 苯并三唑的FVP反应Fig.38 FVP reaction of benzotriazole
同样地,CHOU等把底物分子依托酸酐在FVP条件下反应,分子内脱去一分子的二氧化碳后再关环最终形成氮杂四元环化合物,其产率达到80%,该不稳定的产品需要在-90 ℃条件下被核磁谱表征[52](图39).
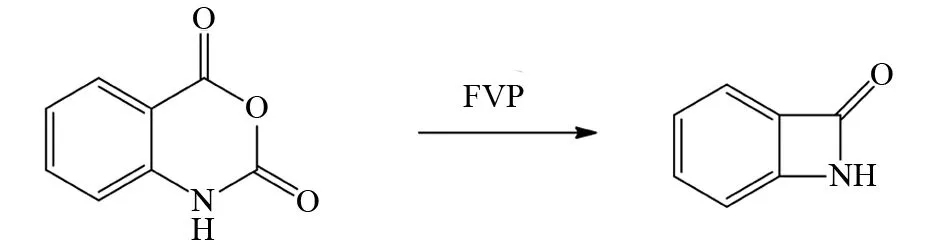
图39 依托酸酐的FVP反应Fig.39 FVP reaction of isatoic anhydride
MEIER等通过类似的方法可获得硫杂四元环化合物,底物分子邻羟甲基萘硫酚在FVP条件分子内脱去一分子的水后最终生成四元硫杂环化合物[53](图40).
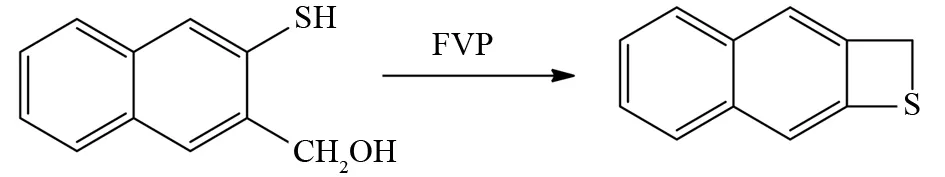
图40 邻羟甲基萘硫酚的FVP反应Fig.40 FVP reaction of (3-mercaptonaphthalen-2-yl)methanol
BETTINGER等把硼氮杂苯的氯化衍生物通过以二氧化碳作为载气的FVP反应,反应底物先脱去氯化硅烷分子形成1,2-azaborinine中间体,这个中间体非常活泼,只能在温度为12 K以下的情况下稳定. 在FVP条件下,甚至连性质相对稳定的二氧化碳气体分子都可以与其加成形成四元B,N杂环化合物[54]. 当然该四元杂环化合物也不太稳定,其在光照条件下便可开环生成新的中间体(图41).
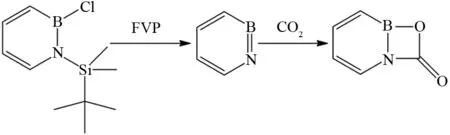
图41 硼氮杂苯衍生物的FVP反应Fig.41 FVP reaction of boron azatriphenylene derivative
4 结论
本文介绍了FVP方法的发展背景和研究进展,从实验装置、反应机理等角度出发,着重列举了FVP方法合成各类有机化合物的应用实例. 和传统的有机合成方法相比,FVP方法不依赖反应溶剂,可通过高温提供足够的反应能量,而低温收集又有利于稳定性质活泼的合成产物. 总体来说,FVP为有机合成反应提供了一种独特的方法,尤其适用于一些常规有机合成方法难以获得的反芳香性化合物、四元杂环化合物、以及具有较大张力的碗状或笼状结构的化合物的合成. 但该方法多数情况下只适合分子内反应,也不适用于一些在真空条件都难以挥发的底物,使FVP方法在底物选择上受到一定的限制. 相信随着FVP反应技术的发展,将进一步突破反应底物的局限,并从分子内反应向双分子或多分子反应延伸,使热解方法在有机化学领域的应用得到拓展.
[1] HURD C D. The pyrolysis of carbon compounds [M]. Chem Eng News, 1929.
[2] GOLDEN D M, SPOKES G N, BENSON S W. Very low-pressure pyrolysis (VLPP): A versatile kinetic tool[J]. Angew Chem Int Ed, 1973, 12(7): 534-546.
[3] KARPF M. Organic synthesis at high temperatures. Gas-phase flow thermolysis [J]. Angew Chem Int Ed, 1986, 25(5): 414-430.
[4] MCNAB H. Synthetic app-lications of flash vacuum pyrolysis [J]. Contemp Org Synth, 1996, 3(5): 373-396.
[5] MCNAB H. Chemistry without reagents: Synthetic applications of flash vacumpyrolysis [J]. Aldrichim Acta, 2004, 37(1): 19-26.
[6] DUFFY E F, FOOT J S, MCNAB H, et al. An empirical study of the effect of the variables in a flash vacuum pyrolysis (FVP) experiment [J]. Org Biomol Chem, 2004, 2(18): 2677-2683.
[7] HICKSON C L, MCNAB H. Furo[3,2-b]pyridine and thieno[3,2-b]pyridine [J]. Synthesis, 1981, 9(6): 464-465.
[8] WIERSUM U E, MIJS W J. Preparative flash vacuum thermolysis. A retro Diels-Alder reaction as a convenient route to isobenzofuran [J]. J Chem Soc, Chem Commun, 1972(6): 347-348.
[9] BOWIE R, GARDNER D, MCOMIE J, et al. The pyrolysis of polycarbonyl compounds. II. Synthesis of biphenylenes from phthalic anhydrides and other carbonyl compounds [J]. Aust J Chem, 1967, 20(1): 139-148.
[10] CADOGAN J, HICKSON C L, MCNAB H. Short contact time reactions of large organic free radicals [J]. Tetrahedron, 1986, 42(8): 2135-2165.
[11] BLACK M, CADOGAN J, MCNAB H. Pyrolysis ofo-ally1 salicylic amides and esters, and related compounds: Formation of lsoindolones and phthalides [J]. J Chem Soc Perkin, 1994(2): 155-159.
[12] TRAHANOVSKY W S, ONG C C, LAWSON J A. Organic oxalates. II. Formation of bibenzyls by pyrolysis of benzyl oxalates [J]. J Am Chem Soc, 1968, 90(11): 2839-2842.
[13] LEONARD E C. Pyrolysis of dibenzylsulfones [J]. J Org Chem, 1962, 27(5): 1921-1923.
[14] BROWN R, EASTWOOD F, HARRINGTON K, et al. Methyleneketenes and methylenecarbenes. III. Pyrolyticsynthesis of arylacetylenes and their thermal rearrangements involving arylrnethylenecarbenes [J]. Aust J Chem, 1974, 27(11): 2391-2402.
[15] VÖGTLE F, ROSSA L. Pyrolysis of sulfones as a synthetic method [J]. Angew Chem Int Ed, 1979, 18(7): 515-529.
[16] SEKINE Y, BOEKELHEIDE V. A study of the synthesis and properties of [26](1,2,3,4,5,6)cyclophane (superphane) [J]. J Am Chem Soc, 1981, 103(7): 1777-1785.
[17] BOEKELHEIDE V. [2n]Cyclophanes: paracyclophane to superphane [J]. Acc Chem Res, 1980, 13(3): 65-70.
[18] RÜEDI G, HANSEN H J. Cyclopropylcarbinylradicals as three-carbon insertion units: easy synthesis of C-15 macrocyclicketones by three-carbon ringexpansion [J]. Tetrahedron Lett, 2004, 45(26): 5143-5145.
[19] RIPOLL J L, VALLÉE Y. Synthetic applications of the retro-enereaction [J]. Synthesis, 1993, 1993(7): 659-677.
[20] JENNESKENS L W, HOEFS C, WIERSUM U. Preparative flash vacuumthermolysis. Selective elimination of 6-chloro-1-hexene from esters of 6-chloro-1-hexanol with schoenberg rearrangement of the s-methyl xanthate [J]. J Org Chem, 1989, 54(24): 5811-5814.
[21] SAUER N N, ANGELICI RJ, HUANG Y, et al. A convenient synthesis of 2,3-dihydrothiophene [J]. J Org Chem, 1986, 51(1): 113-114.
[22] KLUNDER A, ZHU J, ZWANENBURG B. The concept of transient chirality in the stereoselective synthesis of functionalized cycloalkenes applying the retro-Diels-Alder methodology [J]. Chem Rev, 1999, 99(5): 1163-1190.
[23] MANDVILLE G, GIRARD C, BLOCH R. A general and stereoselective approach to nonactateesters and isomers: a versatile synthesis of (+)-methyl nonactate [J]. Tetrahedron, 1997, 53(50): 17079-17088.
[24] MEHTA G, SRIKRISHNA A, REDDY AV, et al. A novel, versatile synthetic approach to linearly fused tricyclopentanoids via photo-thermal olefin metathesis [J]. Tetrahedron, 1981, 37(25): 4543-4559.
[25] BRAIN PT, SMART BA, ROBERTSON HE,et al. Molecular structure of 3,4-dimethylenehexa-1,5-diene ([4]dendralene), C8H10, in the gas phase as determined by electron diffraction andabinitio calculations [J]. J Org Chem, 1997, 62(9): 2767-2773.
[26] TRAHANOVSKY W S, CASSADY T J, WOODS T L. Preparation of 2,3-dimethylene-2,3-dihydrofurans by the flash vacuum pyrolysis of substituted furylmethylesters [J]. J Am Chem Soc, 1981, 103(22): 6691-6695.
[27] SCHIESS P, HEITZMANN M. Hexakis (methylidene)-cyclohexane (“[6]Radialene”). Chemical and spectralproperties [J]. Helv Chim Acta, 1978, 61(2): 844-847.
[28] RIPOLL J L, THUILLIER A. Retrodienic reactions V. Cumulenes by flash thermolysis of 9,10-ethano-9,10-dihydroanthracenes. Synthesis of pentatetraene [J]. Tetrahedron, 1977, 33(11): 1333-1336.
[29] RIPOLL J L, ROUESSAC A, ROUESSAC F. Recent applications of the retro-Diels-Alder reaction in organic synthesis [J]. Tetrahedron, 1978, 34(1): 19-40.
[30] HUNTER G A, MCNAB H. Phenylthioacetylene [J]. Synthesis, 1993(11): 1067-1068.
[31] WENTRUP C, WINTER H W. A General and facile synthesis of aryl- and hetero-arylacetylenes [J]. Angew Chem Int Ed, 1978, 17(8): 609-610.
[32] AITKEN R A, ATHERTON J I. Flash vacuum pyrolysis of stabilised phosphorus ylides. Part 1. Preparation of aliphatic and terminal alkynes [J]. J Chem Soc Perkin 1, 1994(10): 1281-1284.
[33] SCOTT L T, BOORUM M M, MCMAHON B J, et al. A rational chemical synthesis of C60[J]. Science, 2002, 295(5559): 1500-1503.
[34] BROWN R, MCMULLEN G. Methyleneketenes and methylenecarbenes. II. A new phenolic ring synthesis: 2-naphthol from o-tolualdehyde [J]. Aust J Chem, 1974, 27(11): 2385-2391.
[35] BROWNR, JONES C. Methyleneketenes and methylenecarbenes. XV. A new synthesis of ruscodibenzofuran [J]. Aust J Chem, 1980, 33(8): 1817-1822.
[36] KOLLER M, KARPF M, DREIDING A S. The gas-flow thermolysis of 1-isobutenyl alkynyl and 2-methylphenyl alkynyl ketones. Synthesis of methylenomycin B [J]. Helv Chim Acta, 1986, 69(3): 560-579.
[37] ALDER R W, EAST SP, HARVEY J N, et al. The azulene-to-naphthalene rearrangement revisited: a DFT study of intramolecular and radical-promoted mechanisms [J]. J Am Chem Soc, 2003, 125(18): 5375-5387.
[38] MACBRIDE J. Formation of biphenylenes by thermal extrusion of molecular nitrogen from benzo[c]cinnolines [J]. J Chem Soc Chem Commun, 1972, (22): 1219-1220.
[39] JOINES R C, TURNER A B, JONES W M. The Rearrangement of phenylcarbenes to cycloheptatrienylidenes [J]. J Am Chem Soc, 1969, 91(27): 7754-7755.
[40] MEHTA G, RAO H S P. Synthetic studies directed towards bucky-balls and bucky-bowls [J]. Tetrahedron, 1998, 54(44): 13325-13370.
[41] BRONSTEIN H E, CHOI N, SCOTT L T. Practical synthesis of an open geodesic polyarene with a fullerene-type 6:6-double bond at the center: diindeno[1,2,3,4-defg;1′,2′,3′,4′-mnop]chrysene [J]. J Am Chem Soc, 2002, 124(30): 8870-8875.
[42] BROOKS M A, SCOTT L T. 1,2-Shifts of hydrogen atoms in aryl radicals [J]. J Am Chem Soc, 1999, 121(23): 5444-5449.
[43] SAROBE M, KWINT H C, FLEER T, et al. Flash vacuum thermolysis of acenaphtho[1,2-a]acenaphthylene, fluoranthene, benzo[k]- and benzo[j]fluoranthene-homolytic scission of carbon-carbon single bonds of internally fused cyclopenta moieties att≥ 1 100 ℃ [J]. Eur J Org Chem, 1999, 1999(5): 1191-1200.
[44] SCOTT L T, HASHEMI M M, MEYER D T, et al. Corannulene. A convenient new synthesis [J]. J Am Chem Soc, 1991, 113(18): 7082-7084.
[45] SCOTT L T, CHENG P C, HASHEMI M M, et al. Corannulene. A three-step synthesis1 [J]. J Am Chem Soc, 1997, 119(45): 10963-10968.
[46] IMAMURA K, TAKIMIYA K, OTSUBO T, et al. Triphenyleno[1,12-bcd:4,5-b′c′d′:8,9-b″c″d″]trithiophene: the first bowl-shaped heteroaromatic [J]. Chem Commun, 1999(18): 1859-1860.
[47] SCOTT L T, JACKSON E A, ZHANG Q, et al. A short, rigid, structurally pure carbon nanotube by stepwise chemical synthesis [J]. J Am Chem Soc, 2012, 134(1): 107-110.
[48] AMSHAROV K Y, JANSEN M. A C78fullerene precursor: toward the direct synthesis of higher fullerenes [J]. J Org Chem, 2008, 73(7): 2931-2934.
[49] AMSHAROV K Y, JANSEN M. Synthesis of a higher fullerene precursor-an “unrolled” C84fullerene [J]. Chem Commun, 2009(19): 2691-2693.
[50] GABER A, MCNAB H. Synthetic applications of the pyrolysis of Meldrum’s acid derivatives [J]. Synthesis, 2001, 2001(14): 2059-2074.
[51] MCNAB H, WITHELL K. Synthesis of 2-substituted 1,3-oxazin-6-ones by gas-phasepyrolysis [J]. Tetrahedron, 1996, 52(9): 3163-3170.
[52] CHIU S, CHOU C H. Facile synthesis of reactive benzoazetinone by flash vacuum pyrolysis of isatoicanhydride [J]. Tetrahedron Lett, 1999, 40(52): 9271-9272.
[53] MEIER H, RUMPF N. Angular benzobisthietes [J]. Tetrahedron Lett, 1998, 39(52): 9639-9642.
[54] EDEL K, BROUGH S A, LAMM A N, et al. 1,2-Azaborine: the boron-nitrogen derivative ofortho-benzyne [J]. Angew Chem Int Ed, 2015, 54(27): 7819-7822.
[责任编辑:吴文鹏]
Flash vacuum pyrolysis (FVP) method applied in organic synthesis
XU Piaoyang1, ZHANG Qianyan2, XIE Suyuan2, GAO Fei1*
(1.CollegeofChemistryandEnvironment,MinnanNormalUniversity,Zhangzhou361000,Fujian,China;2.CollegeofChemistryandChemicalengineering,XiamenUniversity,Xiamen361005,Fujian,China)
FVP (Flash vacuum pyrolysis) is a pyrolysis reaction process occurred when reactant evaporated or sublimed under vacuum then rapidly passes through high temperature zone, which is often used to synthesize some important nonplanar aromatic compounds, such as the famous corannulene(C20H10), fullerene C60and so on.This paper reviewed the development history of the FVP method, basic structure of FVP apparatus, reaction mechanism of FVP reaction and practical application in organic synthesis. Compared with the traditional organic synthesis, the advantage of the FVP method is that it can provide the higher external energy to help the formation of chemical bonds and a more rapid cooling way to help stabilize the resulting product. Therefore, FVP method can be employed synthesizing some thermodynamic unstable products, as well as some target compounds which are difficult to be obtained by the conventional organic synthesis method. On the other hand, the FVP method has its limitations, which is not applicable for some reactants to difficult volatilize under vacuum and ordi- nary bimolecular reactions and multi-molecular reactions inorganic synthesis, because FVP reaction processes in gas phase and intramolecular elimination reaction usually dominates. As a unique and practical organic synthetic method, FVP has been widely used and continuously developed in organic synthesis.
FVP method;organic synthesis;gas phase synthesis;intramolecular reaction
1008-1011(2016)04-0523-14
2016-01-17.
国家自然科学基金项目(21301143).
徐飘扬(1991-),男,硕士生,研究方向为碳原子簇化学.*通讯联系人,E-mail:fgao@mnnu.edu.cn.
O621.3
A
