LC-MS/MS 测定小鼠肝脏中甘草次酸含量
2016-08-16王守云霍韬光冯雪松
王守云, 霍韬光, 封 聪, 姜 泓, 冯雪松*
(1.中国医科大学 药学院,辽宁 沈阳 110122; 2.中国医科大学 公共卫生学院,辽宁 沈阳 110122)
LC-MS/MS 测定小鼠肝脏中甘草次酸含量
王守云1, 霍韬光2, 封聪2, 姜泓2, 冯雪松1*
(1.中国医科大学 药学院,辽宁 沈阳 110122;2.中国医科大学 公共卫生学院,辽宁 沈阳 110122)
建立了LC-MS/MS测定小鼠肝脏中甘草次酸含量的方法. 本法以咖啡酸为内标物质,采用多反应监测模式,对肝脏中甘草次酸进行含量测定. 所建方法灵敏、准确、可靠,具有线性范围宽、专属性强、基质效应低、回收率及稳定性高的特点,可用于甘草次酸与雄黄联合用药小鼠肝脏中甘草次酸的含量测定.
LC-MS/MS;甘草次酸;咖啡酸;多反应监测;肝脏
甘草次酸(Glycyrrhetinic acid, GA)为甘草在体内的主要代谢产物,具有抗肿瘤、抗炎抗菌、治疗心血管疾病、免疫调节、抗HIV及肾上腺皮质激素样作用等多种药理作用,能够增加肝肾解毒功能[1-2]. 临床上,甘草常与雄黄配伍使用,研究表明甘草次酸对雄黄具有“解毒”作用[3-4].
目前,采用液相色谱-质谱法(LC-MS/MS)测定血浆中甘草酸盐或甘草次酸浓度的测定方法已有文献报道[5-7],常选用白藜芦醇、丙酸睾酮及甘草次酸甲酯等作为内标物质,而采用咖啡酸作为内标物质检测肝脏中甘草次酸含量的研究鲜有报道. 本研究将选用咖啡酸为内标物,采用多反应监测模式建立LC-MS/MS测定小鼠肝脏中甘草次酸含量的方法,并将本法用于甘草次酸与雄黄联合用药小鼠肝脏中甘草次酸的含量测定,为探讨甘草与雄黄配伍机制的研究提供研究思路.
1 实验部分
1.1仪器与试剂
1290型高效液相色谱仪(Agilent公司,美国),6410型三重串联四级杆质谱仪(Agilent公司,美国),CP124C全自动电子天平(十万分之一)(日本A&D公司);3K-18型超速低温冷冻离心机(Sigma公司);超声波细胞粉碎机(宁波新芝生物科技股份有限公司);XW-80A旋涡混合器(上海精科实业有限公司);真空离心浓缩仪(湖南赫西仪器装备有限公司);Easypure纯水系统(Barnstead公司,美国).
雄黄(产地,甘肃),甘草次酸(上海源叶生物科技有限公司),咖啡酸(中国药品生物制品检定所),质谱级乙腈(Sigma公司),质谱级甲醇(Sigma公司),其他试剂均为分析纯.
1.2实验方法
1.2.1动物处理
SPF级雄性ICR小鼠50只,体重(20±5) g,均由中国医科大学实验动物部提供. 标准饲料喂养,小鼠自由摄食及饮水,实验前适应性饲养1 w. 按照体重随机分为5组,每组10只. 第1组为对照组,灌胃给予0.5%羧甲基纤维素钠(CMC-Na)水溶液;第2组为雄黄组,灌胃给予雄黄(1.35 g/kg);第3组为甘草次酸对照组,灌胃给予甘草次酸48 mg/kg;第4组为甘草次酸低剂量联合用药组,灌胃给予甘草次酸(16 mg/kg)+雄黄(1.35 g/kg);第5组为甘草次酸高剂量联合用药组,灌胃给予甘草次酸(48 mg/kg)+雄黄(1.35 g/kg). 每日灌胃1次,连续8 w. 每隔3 d称量体重1次,调节灌胃容量. 于末次给药24 h后,无水乙醚麻醉,冰浴取小鼠肝脏组织,预冷0.9%生理盐水冲洗干净,待用.
1.2.2甘草次酸标准溶液和内标溶液的配制
精密称取甘草次酸对照品适量,用甲醇溶解,定容至100 mL,得浓度为336.0 mg/L的甘草次酸储备液,备用. 精密吸取储备液适量,稀释成浓度为67.20、134.4、336.0、672.0、1 344、6 720 μg/L的标准系列液.
精密称取咖啡酸对照品适量,用甲醇溶解,定容至100 mL,得浓度为900.0 μg/L的咖啡酸储备液,备用. 临用前,精密量取储备液适量,稀释成浓度为630 .0 μg/L的内标溶液.
1.2.3样品溶液的制备
精密称取小鼠肝脏200 mg,加入乙腈1.0 mL,超声破碎2 min,得20%(质量体积比)的肝组织匀浆液,然后向匀浆液中加入浓度为630.0 μg/L咖啡酸溶液200 μL,混匀,4 ℃离心10 min(12 000 r·min-1). 精密吸取上清液1.0 mL,浓缩至干,残渣以乙腈-水(80∶20,体积比)50 μL复溶后,4 ℃离心(2 000 r·min-1)5 min ,取上清液,进样分析.
1.2.4色谱条件和质谱条件
ZORBAX SB-C18色谱柱(Agilent,4.6×100 mm,3.5 μm),柱温30 ℃,流动相为乙腈-水(80∶20,体积比),流速为1 mL/min,进样量为10 μL.
电喷雾(ESI)离子源,负离子模式,采用多反应监测(MRM)方式,甘草次酸m/z为469.2→425.2,咖啡酸m/z为179.1→135.1. 毛细管电压为3.5 kV,甘草次酸和咖啡酸的裂解电压分别为200 V和110 V,碰撞电压分别为38 V和15 V. 雾化器压力为275.792 kPa,离子源温度为350 ℃,保护气流量为10 L/min.
1.2.5样品测定
按“1.2.3”项下方法操作,测定对照组、雄黄组、甘草次酸对照组、甘草次酸低剂量联合用药组及甘草次酸高剂量联合用药组小鼠肝脏组织中甘草次酸的含量,并以当日随行的标准曲线计算小鼠肝脏组织中甘草次酸的浓度.
1.2.6统计分析
所得数据以平均值±标准差表示,用SPSS 17.0软件单因素方差分析方法(ANOVA)进行各指标组间差异的显著性检验,以P< 0.05作为检验的显著性差异.
2 结果与讨论
2.1内标物的选择
在内标物的选择过程中,考查了与甘草次酸结构类似的齐墩果酸、熊果酸、熊去氧胆酸及咖啡酸. 实验结果表明,咖啡酸与待测物受内源物质干扰及试剂、仪器等因素的影响相近,且色谱分离时间短,出峰时间仅为0.927 min. 甘草次酸及内标物咖啡酸在MRM监测中信号无相互干扰,最终选择咖啡酸作为内标物.
2.2色谱及质谱条件的优化
乙腈-水和甲醇-水作为反相液相色谱常用的流动相,对待测分析物的色谱保留时间有较大影响,且流动相的组成与分离度及待测物的离子化程度有关. 实验过程中,为了使分析方法的灵敏度更高,考察了两个流动相体系对它们的影响. 实验结果表明,使用乙腈-水作为流动相的色谱峰形对称,且化合物出峰时间快,总体分析时间短. 实验又探讨了流动相中加入不同浓度(体积比0.1%、0.2%)甲酸及不加甲酸对待测物的灵敏度及色谱峰形的影响,结果表明甲酸对色谱峰形及灵敏度并无明显影响. 最终,流动相选择乙腈-水(80∶20,体积比).
结果显示,负离子检测较正离子检测模式信号强度大,最终选择负离子检测模式. 对甘草次酸及内标物咖啡酸进行一级全扫描,甘草次酸和咖啡酸的分子离子峰[M-H]-m/z分别为469.2和179.1,选择其作为母离子进行二级全扫描,最终选择m/z为469.2→425.2及179.1→135.1分别为甘草次酸和咖啡酸MRM分析的定量离子对.
在质谱条件优化中,毛细管出口电压(fragmentor voltage)及碰撞能(collision energy)对信号影响显著. 因此在本文中仔细考察了毛细管出口电压在15~150 V之间及碰撞能在5~25 V之间对甘草次酸和咖啡酸信号强度的影响. 结果表明,在200 V毛细管出口电压、38 V碰撞能下,能够得到离子强度合适的甘草次酸子离子;在110 V毛细管出口电压、15 V碰撞能下,能够得到离子强度合适的咖啡酸离子.
2.3方法的专属性
由图1可见,甘草次酸和咖啡酸的保留时间分别为3.265和0.927 min,咖啡酸可作为测定甘草次酸含量的内标物质. 小鼠肝组织中其他内源性物质不干扰甘草次酸和咖啡酸的分离. 结果表明,该方法具有良好专属性.
2.4标准曲线和线性范围、精密度、准确度、基质效应及回收率
取6只对照组小鼠混合肝组织匀浆液,分别加入甘草次酸系列对照品溶液和咖啡酸内标溶液,在优化的实验条件下,利用建立的LC-MS/MS联用系统进行分析,采用峰面积积分. 以甘草次酸和咖啡酸内标物色谱峰面积比值为纵坐标,以甘草次酸浓度为横坐标,采用加权最小二乘法线性回归,求得回归方程. 小鼠肝脏组织中甘草次酸的线性回归方程为y=0.113 61x+1.703 5(r=0.999 8),结果表明甘草次酸在9.600~960.0 μg/L范围内线性关系良好.

A.对照组色谱图;B.甘草次酸标准色谱图;C.甘草次酸高剂量联合用药组色谱图.图1 甘草次酸肝脏样品的LC-MS/MS色谱图Fig.1 LC-MS/MS chromatogram of Glycyrrhetinic acid in liver samples
在优化的实验条件下,由表1可见,方法的精密度、准确度及回收率较好,基质效应较低. 低、中、高三个浓度的质控(QC)样品的日内、日间精密度在0.1%~4.1%之间,准确度在0.1%~6.7%之间. 甘草次酸的基质效应在103.5%~104.2%之间(RSD在0.8%~5.3%之间),咖啡酸的基质效应为95.27%(RSD=7.2%). 草次酸的提取回收率在97.1%~102.4%之间,内标物咖啡酸的提取回收率为104.96%(RSD=7.6%).

表1 小鼠肝脏组织中甘草次酸测定方法的精密度、准确度、基质效应及回收率
2.5样品稳定性考察结果
在优化的实验条件下,三个浓度的QC样品,经3次冷冻-解冻循环后(-20~20 ℃)、室温放置24 h及-20 ℃储存30 d. 由表2可见稳定性良好.
2.6应用及讨论
结果表明,甘草次酸对照组、甘草次酸低剂量联合用药组、甘草次酸高剂量联合用药组中甘草次酸的含量分别为(3 730.4±1 016.2) ng/g、(317.0±188.9) ng/g和(1 158.4±176.1) ng/g,对照组未检出甘草次酸. 甘草次酸高剂量组、甘草次酸对照组与甘草次酸低剂量组比较有统计学意义(P<0.05).
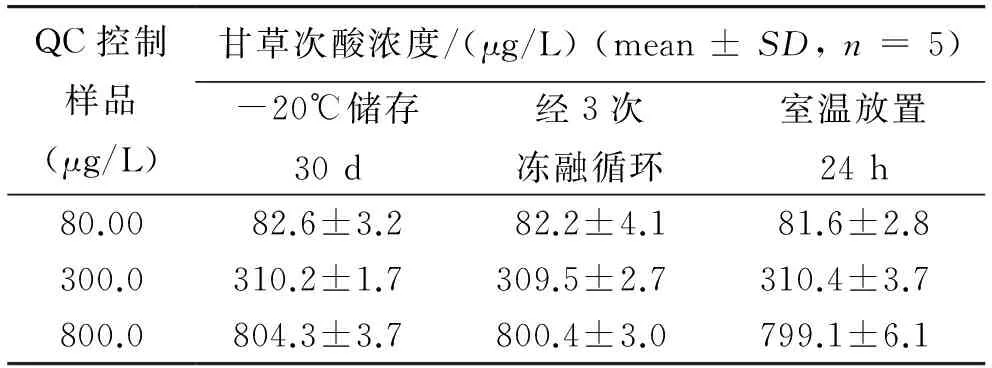
表2 小鼠肝脏组织中甘草次酸的稳定性考察
与甘草次酸对照组比较,甘草次酸联合用药组小鼠肝脏中甘草次酸的含量明显降低(P<0.05),甘草次酸与雄黄高剂量联合用药组的甘草次酸含量明显高于甘草次酸低剂量联合用药组(P<0.05).
雄黄的毒性源于雄黄中的砷可被机体吸收,并在体内(如脑、肝脏、肾脏)蓄积[8-11],而长期服用含雄黄制剂易引起药源性慢性砷中毒,造成肝脏损害[12]. 临床上,雄黄常与甘草配伍使用. 甘草为国老药,具有调和诸药的作用,对雄黄的毒性也有一定的解毒作用[3]. 本研究表明,甘草次酸与雄黄联合用药后,肝脏中甘草次酸含量明显降低,可能是砷与甘草次酸结合,使肝脏中游离态的甘草次酸含量减少,这可能也是甘草次酸对雄黄起解毒作用的原因之一,具体机制有待进一步探讨.
3 结论
采用LC-MS/MS法,以咖啡酸为内标物质,通过MRM负离子模式监测,对甘草次酸与雄黄联合用药组小鼠肝脏组织中的甘草次酸进行含量测定,建立了肝脏中甘草次酸的含量测定方法,为甘草与雄黄配伍机制的研究提供实验数据.
[1] 刘彬, 齐云. 甘草酸甘草次酸的药理学研究进展[J]. 国外医药:植物药分册, 2006, 21(3): 100-103.
[2] 张明发, 沈雅琴. 甘草酸及其苷元甘草次酸的糖皮质激素样作用[J]. 现代药物与临床, 2011, 26(1): 33-35.
[3] 董菊, 严晓鹰, 王明燕, 等. 牛黄解毒片配伍影响雄黄砷毒性的动物实验研究[J]. 时珍国医国药, 2014, 2(25): 317-319.
[4] 何丹, 刘凤琴, 李焕德. 甘草解毒作用研究进展[J]. 中南药学, 2009, 7(12): 927-931.
[5] ZHAO W J, WANG B J, WEI C M, et al. Determination of glycyrrhetic acid in human plasma by HPLC-MS method and investigation of its pharmacokinetics [J]. J Clin Pharm Ther, 2008, 33(3): 289-294.
[6] LIN Z P, QIU S X, WUFUER A, et al. Simultaneous determination of glycyrrhizin, a marker component in Radix Glycyrrhizae, and its major metabolite gylcyrrhetic acid in human plasma by LC-MS/MS [J]. J Chromatogr B, 2005, 814(2): 201-207.
[7] 黄鑫, 丁黎, 杨劲, 等. 高效液相色谱-质谱法研究甘草酸二铵胶囊在健康人体的药代动力学[J]. 中国临床药理学杂志, 2006, 22(5): 365-368.
[8] 姜泓, 张颖花, 丁敬华, 等. HPLC-HG-AFS法测定雄黄中As(III)的含量[J]. 化学研究, 2008, 19(4): 67-69.
[9] 张颖花, 霍韬光, 姜泓, 等. 高效液相色谱-氢化物发生-原子荧光光谱法检测牛黄解毒片中的砷[J]. 化学研究, 2012, 23(4): 60-63.
[10] 苑洁, 霍韬光, 王艳蕾, 等. HG-FAAS法测定雄黄染毒小鼠肝及肾脏中的砷含量[J]. 化学研究, 2014, 25(1): 82-85.
[11] 张颖花, 高咏, 霍韬光, 等. 利用氢化物发生-冷阱捕集-原子吸收光谱联用技术测定雄黄染毒大鼠血中砷的含量[J]. 化学研究, 2013, 24(3): 274-276.
[12] 张秀娟, 白雪莹, 陆童, 等. 雄黄对小鼠肝脏功能损伤的初步研究[J]. 中药药理与临床, 2014, 30(1): 56-58.
[责任编辑:吴文鹏]
Determination of glycyrrhetinic acid in liver of mice by LC-MS/MS
WANG Shouyun1, HUO Taoguang2, FENG Cong2, JIANG Hong2, FENG Xuesong1*
(1.CollegeofPharmacy,ChinaMedicalUniversity,Shenyang110122,Liaoning,China;2.CollegeofPublicHealth,ChinaMedicalUniversity,Shenyang110122,Liaoning,China)
A method of LC-MS/MS was established for the determination of glycyrrhetinic acid in liver of mice. Multiple reaction monitoring(MRM) scanning mode was used for the quantification with caffeic acid as the internal standard (IS). The method was validated for sensitivity, precision, reliability, a wide linear range, strong specificity, low matrix effect, high recovery and stability. The method was successfully applied for the determination of glycyrrhetinic acid in liver of mice combined treated with glycyrrhetinic acid and realgar.
LC-MS/MS; glycyrrhetinic acid; caffeic acid; multiple reaction monitoring; liver
1008-1011(2016)04-0429-04
2016-01-11.
国家自然科学基金(81473417, 81403066).
王守云(1991-), 女, 硕士生, 研究方向为药物分析.*通讯联系人, E-mail: voncedar@126.com.
R286.02
A
