药物性肝损伤的代谢、遗传学机制
2016-08-10王书杰李晓天张莉蓉
王书杰,王 沛,李晓天,张莉蓉
(郑州大学 1.药学院药理学系;2.基础医学院药理学系,河南 郑州 450001)
药物性肝损伤的代谢、遗传学机制
王书杰1,2,王沛2,李晓天1,张莉蓉2
(郑州大学 1.药学院药理学系;2.基础医学院药理学系,河南 郑州450001)
药物性肝损伤;肝毒性;代谢;遗传;白细胞抗原;CYP450
1 前言
肝脏是大多数药物进行生物转化的场所。很多药物在体内发挥治疗作用的同时,不可避免地会影响肝脏的结构与功能,导致各种类型的药物性肝损伤(drug-induced liver injury, DILI)。DILI是一类复杂的不良反应,因药物本身或/及其代谢产物,或因特殊体质对药物的超敏感性或耐受性降低,导致的肝脏功能异常或肝脏损伤。
据统计,目前有超过1 100种药物可引起DILI。常见的有对乙酰氨基酚(acetaminophen, APAP)、异烟肼、利福平、氟氯西林、苯妥英及中草药等。

Tab 1 Drugs commonly causing liver damage
大量研究表明,DILI的发生与药物、药物活性代谢产物、代谢酶和转运体活性、胆汁淤积、线粒体功能障碍及氧化应激等多种因素有关,已证实性别和体重与肝毒性有明显相关性,环境因素如酶诱导、酒精、营养不良可能是引发DILI的一系列因素[1-2]。
尽管几乎所有类别的药物均可引起DILI,但DILI仍是一种罕见的不良反应。本文所指的“药”是指成人常用药物。1998年,著名学者将DILI定义为局部性问题,并相继明确了与DILI相关的药物及不良反应。以上有关DILI的人口统计学研究以及在DILI发病机制领域所取得的重大进展,引起了研究人员对DILI的代谢和遗传学基础的关注。
本文将从代谢、遗传两个方面综述成人用药引起DILI的发病机制。虽然单克隆抗体和用来治疗感染性肝炎及癌症的药物都和肝损伤有关,但因这些药物引起的DILI是代谢和遗传两种机制共同作用的结果,因此本文不作讨论。
2 DILI的代谢学基础
一般而言,药物代谢的目的是通过产生极性高、水溶性代谢产物以利于从机体排出。药物的代谢反应通常包括Ⅰ相和Ⅱ相反应。Ⅰ相反应包括氧化、还原和水解反应,Ⅱ相反应即结合反应。参与反应的药物代谢酶有氧化酶(如CYP450)、还原酶、水解酶、结合酶(如葡萄糖醛酸基转移酶UGT)。而肝脏药物代谢酶的活性除了受到正常和病理条件下肝脏血流变化的影响,可能还受到与这些酶相关的基因表达的影响。本节将对代谢在DILI中的作用进行综述。在某些情况下,药物或其代谢物通过进入最近的易受损肝细胞引起肝毒性。该机制的代表性药物是APAP。APAP是常用的非处方解热镇痛药,其引起的肝毒性呈剂量相关性。正常情况下,APAP进入肝脏后,经药物代谢酶CYP3A4、CYP2E1、CYP1A2代谢,产生少量具有生物活性的N-乙酰基-对苯醌亚胺(NAPQI)。当过量服用时,NAPQI在肝脏蓄积,结合亚细胞器,引起氧化应激反应,线粒体功能失调和DNA损伤,最终导致肝细胞坏死或凋亡[3]。郭秋平等[4]动物实验证明,SD大鼠服用高剂量APAP后,会产生抑制CYP450的作用,并引起药物在体内蓄积,加剧药物毒性。此外,还有一些药物可通过其他方式引起剂量相关性的肝毒性。如溴芬酸、环磷酰胺(肝窦内皮细胞的直接损伤)或氨甲喋呤可导致肝细胞坏死引起直接肝毒性;可卡因、苯环己哌啶或烟酸可引起缺血性坏死;胺碘酮可引起脂肪性肝炎[5]。代谢在药物或活性代谢物引起的DILI中有重要作用。该机制主要通过3种途径引起肝损伤。第1种方式是通过干扰细胞关键功能直接损伤肝细胞。如APAP、双氯芬酸、利福平、曲格列酮等。第2种方式是增加肝细胞对细胞因子诱导性损伤的敏感性。这一理论已经用于构建DILI的研究模型中,如细菌内毒素的主要成分脂多糖(lipopolysaccharides,LPS)通过肿瘤坏死因子(tumor necrosis factor, TNF-α),在某些情况下使肝脏敏感增加而导致损伤。肝毒性从中度到重度坏死的模型的构建都是运用这个理论。因此,给予单次量LPS或者单次量胺碘酮[11]、曲伐沙星或氟烷[12]时可引发DILI。随着研究的深入,研究人员发现2个及以上刺激物所导致的器官特异性免疫应答的理念可用于自身免疫动物模型的研究中。因此,以上这些新模型有助于DILI的代谢基础的进一步研究。越来越多的研究表明,半抗原(能导致天然蛋白质不可逆性共价修饰的活性代谢产物)在DILI中具有重要作用。这些共价修饰蛋白能促进相应天然蛋白质的免疫识别。在易感人群中,该过程引发细胞因子驱动的一系列的免疫反应,导致肝毒性,这是引起DILI的第3种方式。如苯妥英治疗癫痫时会引起肝损伤。其代谢物和活性氧的产生、肝脏谷胱甘肽的消耗有关,导致肝细胞中线粒体功能受损[13]。而经CYP2C9代谢形成的活性代谢物5-(p-hydroxyphenyl)和HPPH(5-phenylhydantoin),被进一步氧化形成儿茶酚,在肝脏中形成蛋白加合物引起免疫应答[14]。

Tab 2 Drug-induced hepatotoxicity and its underlying mechanisms
虽然本文重点研究肝代谢对肝毒性发展的作用,但肝外药物代谢(小肠、肾微粒体和血浆酯酶等)对药物活性代谢产物的作用亦不可忽视。要充分了解代谢在DILI中的作用,肝外代谢位点对肝毒性的影响的研究是不可或缺的。
3 DILI的遗传学基础
随着人类基因组计划的完成,全基因组关联研究(genome-wide association studies,GWAS)得以实施。它能识别大量相关基因和多个低频率突变,可对低风险疾病的常见遗传变异进行稳定、有效的鉴别,如对患者进行基因筛选可有效预防DILI的发生。氟氯西林和希美加群就是一个成功的案例,在治疗前筛选人类白细胞抗原(human leucocyte antigen, HLA)基因变异可预防肝毒性。HLA呈高度多态性和共显性,是目前所知人体最复杂的系统。HLA单倍型或者其他遗传标记,可以帮助确定服用某种药物时是否产生毒性。遗传学除了在鉴定HLA单倍型中有重要作用,也可影响Ⅰ相和Ⅱ相酶、转运蛋白的表达而参与DILI的发生发展。本节将围绕DILI的遗传学基础进行综述。

Tab 3 GWAS showing significant roles for CYP450 genes
3.1HLA的遗传多态性有关遗传因素和DILI相关性的研究主要集中在HLA单倍型上。事实上,特定人群对DILI的易感性与HLA基因多态性相关。早期研究发现,药物或其代谢产物与细胞内蛋白共价结合形成复合物,HLA将复合物呈递给T细胞,引起不良T细胞应答,进而导致细胞损伤。另外药物也可直接与HLA相互作用,不需形成共价复合物而导致T细胞应答。其中HLA单倍型与抗生素引起的肝损伤有很强的相关性。最早确定的药物是阿莫西林-克拉维酸。Donaldson等[15]运用GWAS发现,具有HLA-DRB1*1501、HLA-DRB5*0101、HLA-DQB1 *0602单倍型的患者服用阿莫西林-克拉维酸产生持续性肝毒性的风险增加10倍。一项大样本量临床试验证实服用阿巴卡韦前进行HLA B*5701检测能有效降低服用阿巴卡韦导致的超敏反应的发生率[16]。Daly等[17]发现,HLA单倍型HLA B*5701位点发生突变的患者服用氟氯西林时肝毒性增加80倍,但HLA B*5701能否用作服用氟氯西林发生DILI的标志物尚需进一步论证。Sharma等[18]发现,具有HLA-DQB1* 0201单倍型的结核患者发生肝毒性的概率增加了1.9倍。遗传图谱的GWAS有助于人们用药的风险评估;但这缺乏统计学显著性研究,加强这些研究可以帮助解释上述结果。
3.2代谢酶的遗传多态性Ⅰ相和Ⅱ相代谢酶的基因表达在DILI的发生发展中具有重要作用[19]。例如CYP2E1*1A 与抗结核病药物的毒性代谢物[20]和活性氧[21]的产生有关;CYP2C8*4与双氯酚酸毒性代谢物产生的肝毒性有关[22]。总之,无论从遗传学角度或是影响疾病过程来讲,均应该考虑CYP450酶表达的差异。
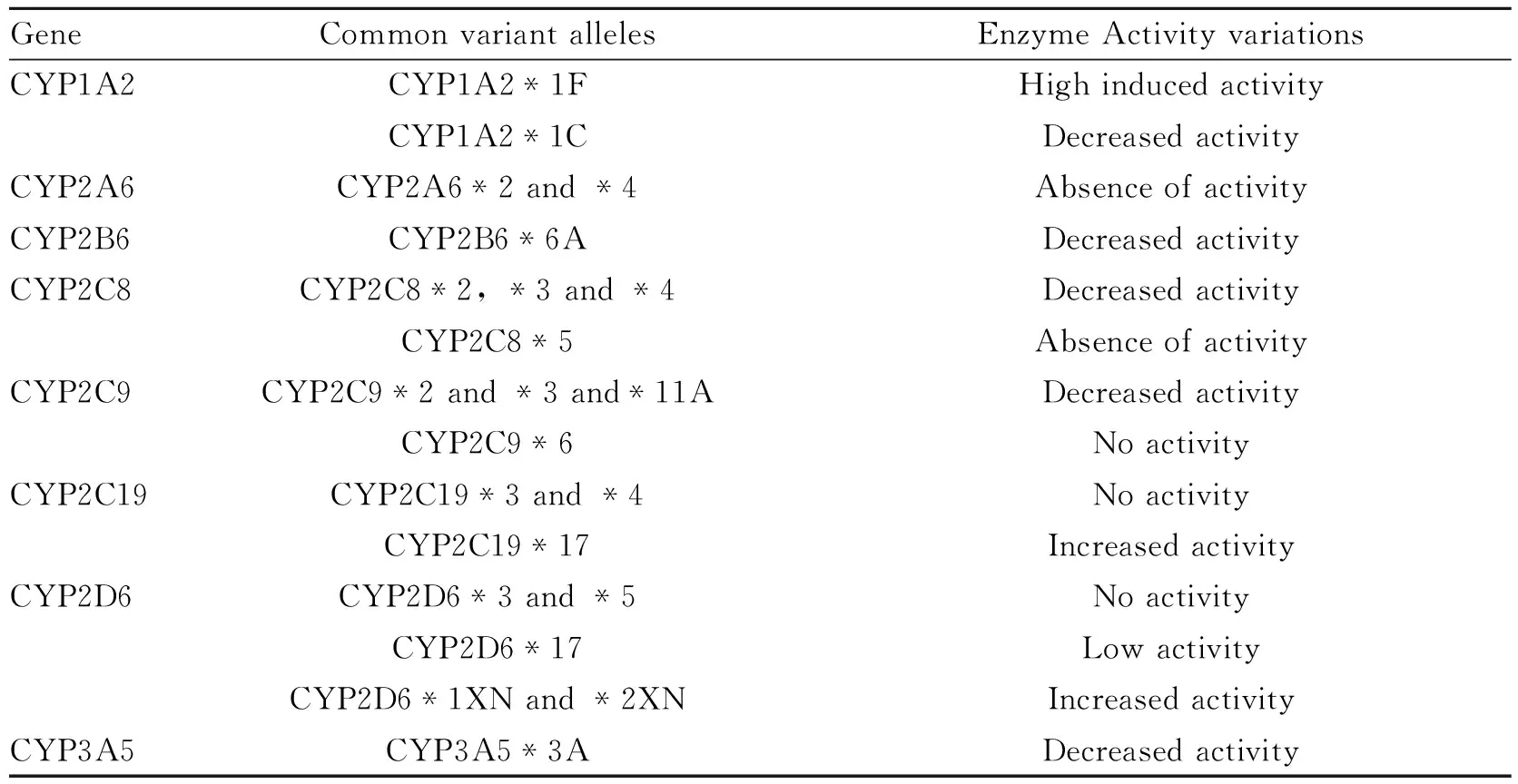
Tab 4 Polymorphisms in cytochromes P450 relevant to drug metabolism and their enzyme activity variations
Only selected alleles and CYP isoforms are shown. Full details are available elsewhere(http:// ww w.cypalleles.ki.se/).
虽然很早有研究表明,Ⅱ相代谢酶与代谢个体差异有关,但在近10年才阐明有关酶差异性表达影响DILI发生的机制。异烟肼的多重代谢产物与药物肝毒性有关。异烟肼经乙酰基转移酶NAT代谢生成的活性代谢物乙酰肼和联氨具有肝毒性。一系列的研究表明,NAT2(慢乙酰化)基因型延迟了异烟肼的毒性代谢产物的解毒[23],是异烟肼引起的肝损伤的危险因素。此外,谷胱甘肽S-转移酶(GST)在异烟肼的活性代谢产物的解毒和活性氧的中和作用中也有重要作用。因此,GSTT1基因缺失型患者服用卡马西平后患中度肝毒性的风险增加[24],在GSTMI或GSTT1基因缺失型患者服用异烟肼后患特异性肝毒性的风险增加[25]。而在GSTM1和GSTT1均是基因缺失型的患者服用曲格列酮后,通过产生活性氧引起线粒体损伤导致ALT水平升高的风险增加。慢乙酰化编码基因NAT2和GSTM1基因携带者发生异烟肼性DILI的概率较快乙酰化和GSTT1基因携带者明显增高[26]。同时,UGT异常时亦能导致肝毒性。Jiang等[27]研究发现,在中国汉族人群中UGT1A9 rs2741045与DILI具有明显相关性。而Daly等[22]发现双氯酚酸的肝毒性与UGT2B 7*2的异常有关,其毒性的机制是产生更多的毒性代谢物。
研究发现抗氧化酶(antioxidant enzymes)能抵御线粒体过氧化物对细胞造成的损伤。其中超氧化物歧化酶(superoxide dismutase, SOD)和谷胱甘肽过氧化酶(glutathione peroxidase, GPx)的遗传多态性与DILI存在关联[28]。Lucena等[29]在研究SOD、GSH-Px时发现,SOD2 rs4880和GPx1 rs1050450突变增加胆汁淤积型或混合型DILI的敏感性。而Kim等[30]研究发现,SOD1 rs2070424明显增加抗结核药物引起肝损伤的风险。
3.3转运蛋白的遗传多态性药物处置相关蛋白的差异性表达,与DILI也有关联。如多药耐药蛋白1(MDR1)和胆盐输出泵(bile salt export pump,BSEP)与DILI有关。其中Ritchie等[31]发现,MDR1 3435 C→T会降低患者服用奈韦拉平和依法韦仑等非核苷逆转录酶抑制药引起肝毒性的风险。而Lang等[32]在研究BSEP和MDR3与肝损伤的关系中发现,BSEP D676Y、G855R和MDR3 I764L突变在胆汁淤积型肝损伤中频率较高,而MDR3 L1082Q突变在细胞损伤型肝损伤频率较高,BSEP 1331 T>C基因突变和BSEP表达降低有关,在胆汁淤积型肝损伤中频率高。
4 结论与展望
据流行病学资料显示,DILI的年发生率为19.5/10万,并呈增高趋势。DILI因具有较高的发病率和死亡率,受到人们的广泛关注。DILI初发症状一般特异性不高,易与基础疾病症状混淆,同时其表征具有一定的隐匿性,部分患者在发生DILI早期肝脏生化指标轻微异常甚至正常。DILI因其不易发现和不可预测性,给诊断和研究造成一定的困难,已经成为亟待解决的问题。
重视和规范DILI的研究是当前刻不容缓的任务。而目前临床上运用传统的肝损伤指标来预测、诊断DILI的能力极为有限。传统肝损伤生物标志物,如ALT、IBIL、AST、ALP、TBIL虽然在对重度DILI的诊断方面占有重要的地位,但在肝损伤的早期诊断、易感人群的识别区分等方面具有一定的局限性。而新型潜在肝损伤生物标志物,如miRNA[33](如miR-122、miR-192)等则还需要进一步的研究评估。未来的目标是寻找更合适的DILI生物标志物及发生DILI风险和预后不良的预测指标,实现可提前识别和筛选DILI的高风险患者。本文围绕代谢、遗传学机制进行研究。迄今为止,虽然关于DILI的机制的研究已有部分成果,但还只是冰山一角,DILI 的发生和进展可能是上述因素相互作用的综合结果。因此,要彻底了解全部的机制,还仍需进一步的研究。
[1]Yuan L Y, Kaplowitz N. Mechanisms of drug-induced liver injury[J].ClinLiverDis, 2013, 17(4): 507-18.
[2]Corsini A, Bortolini M. Drug-induced liver injury:The role of drug metabolism and transport[J].JClinPharmacol, 2013, 53(5): 463-74.
[3]Tujios S, Fontana R J. Mechanisms of drug-induced liver injury: From bedside to bench[J].NatRevGastroHepat, 2011, 8(4): 202-11.
[4]郭秋平,杨威,郭琳,等. 对乙酰氨基酚SD 大鼠毒代动力学研究及P450 的影响[J].中国药理学通报,2014,30(8):1183-4.
[4]Guo Q P, Yang W, Guo L, et al. Study on rats P450 and toxicokinetics induced by APAP[J].ChinPharmacolBull, 2014, 30(8): 1183-4.
[5]Lee W M. Drug-induced hepatotoxicity[J].NewEnglJMed, 2003, 349(20): 474-85.
[6]Boelsterli U A. Diclofenac-induced liver injury: A paradigm of idiosyncratic drug toxicity[J].ToxicolApplPharm, 2003, 192(3): 307-22.
[7]Li F, Lu J, Cheng J, et al. Human PXR modulates hepatotoxicity associated with rifampicin and isoniazid co-therapy[J].NatMed, 2013, 19(4): 418-21.
[8]He K, Talaat R E, Pool W F, et al. Metabolic activation of troglitazone:Identification of a reactive metabolite and mechanisms involved[J].DrugMetabDispos, 2004, 32(6): 639-46.
[9]Lakehal F, Dansette P M, Becquemont L. et al.Indirect cytotoxicity of flucloxacillin toward human biliary epithelium via metabolite formation in hepatocytes[J].ChemResToxicol, 2001, 14(6), 694-701.
[10]Ma L L, Wu Z T, Wang L, et al. Inhibition of hepatic cytochrome P450 enzymes and sodium/bile acid cotransporter exacerbates leflunomide-induced hepatotoxicity[J].ActaPharmacolSin, 2016, 37(3): 415-24.
[11]Lu J T, Jones A D, Harkema J R, et al. Amiodarone exposure during modest inflammation induces idiosyncrasy-like liver injury in rats: Role of tumor necrosis factor-alpha[J].ToxicolSci, 2012, 125(1): 126-33.
[12]Dugan C M, MacDonald A E, Roth R A, Ganey P E. A mouse model of severe halothane hepatitis based on human risk factors[J].JPharmacolExpTher, 2010, 333(2): 364-72.
[13]Eghbal M A, Taziki S, Sattari M R. Mechanisms of phenytoin-induced toxicity in freshly isolated rat hepatocytes and the protective effects of taurine and/or melatonin[J].JBiochemMolToxicol, 2014, 28(3): 111-8.
[14]Sasaki E, Matsuo K, Iida A, et al. A novel mouse model for phenytoin-induced liver injury: involvement of immune-related factors and P450-mediated metabolism[J].ToxicolSci, 2013, 136(1): 250-63.
[15]Donaldson P T, Daly A K, Henderson J, et al. Human leucocyte antigen class II genotype in susceptibility and resistance to co-amoxiclav-induced liver injury[J].JHepatol, 2010, 53(6): 1049-53.
[16]Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 screening for hypersensitivity to abacavir[J].NewEnglJMed, 2008, 358(6): 568-79.
[17]Daly A K, Donaldson P T, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin[J].Nat.Genet, 2009, 41(7): 816-9.
[18]Sharma S K, Balamurugan A, Saha P K, et al. Evaluation of clinical and immunogenetic risk factors for the development of hepatotoxicity during antituberculosis treatment[J].AmJRespCritCare, 2002, 166(7): 916-9.
[19]Daly A K. Genetic polymorphisms affecting drug metabolism: recent advances and clinical aspects[J].AdvPharmacol, 2012, 63:137-67.
[20]Huang Y S, Chern H D, Su W J, et.al. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug-induced hepatitis[J].Hepatology, 2003, 37(4): 924-30.
[21]Vuilleumier N, Rossier M F, Chiappe A, et al. CYP2E1 genotype and isoniazid-induced hepatotoxicity in patients treated for latent tuberculosis[J].EurJClinPharmacol, 2006, 62(6): 423-9.
[22]Daly A K, Aitha G P, Leathart J B, et al. Genetic susceptibility to diclofenac-induced hepatotoxicity: contribution of UGT2B7, CYP2C8, and ABCC2 genotypes[J].Gastroenterology, 2007, 132(1): 272-81.
[23]Metushi I G, Cai P, Zhu X, et al. A fresh look at the mechanism of isoniazid-induced hepatotoxicity[J].ClinPharmacolTher, 2011, 89(6): 911-4.
[24]Ueda K, Ishitsu T, Seo T, et al. Glutathione S-transferase M1 null genotype as a risk factor for carbamazepine-induced mild hepatotoxicity[J].Pharmacogenomics, 2007, 8(5): 435-42.
[25]Leiro V, Fernandez-Villar A, Valverde D, et al. Influence of glutathione S-transferase M1 and T1 homozygous null mutations on the risk of antituberculosis drug-induced hepatotoxicity in a Caucasian population[J].LiverInt, 2008, 28(6): 835-9.
[26]Possuelo L G, Castelan J A, de Brito T C, et al. Association of slow N-acetyltranferase 2 profile and anti-TB drug-induced hepatotoxicity in patients from Southern Brazil[J].EurJClinPharmacol, 2008, 64(7): 673-81.
[27]Jiang J, Zhang X, Huo R, et al. Association study of UGT1A9 promoter ploymorphisms with DILI based on systematically regional variation screen in Chinese population[J].PharmacogenomicsJ, 2015, 15(4): 326-31.
[28]Russmann S, Jetter A, Kullak-Ublick G A,et al. Pharmacogenetics of drug-induced liver injury[J].Hepatology, 2010, 52(2): 748-61.
[29]Lucena M I,Garcia-Martin E, Andrade R J, et al. Mitochondrial superoxide dismutase and glutathione peroxidase in idiosyncratic drug-induced liver injury[J].Hepatology, 2010, 52(1): 303-12.
[30]Kim S H, Kim S H, Lee J H, et al. Superoxide dismutase gene(SOD 1, SOD2, and SOD3) polymorphisms and antituberculosis drug-induced hepatitis[J].AllergyAsthmaImmunolRes, 2015, 7(1): 88-91.
[31]Ritchie M D, Haas D W, Motsinger A A, et al.Drug transporter and metabolizing enzyme gene variants and nonnucleoside reverse-transcriptase inhibitor hepatotoxicity[J].ClinInfectDis, 2006, 43(6): 779-82.
[32]Lang C, Meier Y, Stieger B, et al. Mutations and polymorphisms in the bile salt export pump and the multidrug resistance protein 3 associated with drug-induced liver injury[J].PharmacogenetGenomics, 2007, 17(1): 47-60.
[33]Antoine D J, Dear J W, Lewis P S, et al.Mechanistic biomarkers provide early and sensitive detection of acetaminophen-induced acute liver injury at first presentation to hospital[J].Hepatology, 2013, 58(2): 777-87.
Drug-induced liver injury:metabolic and genetic basis
WANG Shu-jie1,2, WANG Pei2, LI xiao-tian1, ZHANG Li-rong2
(1.DeptofPharmacology,PharmacyCollege; 2.DeptofPharmacology,BasicMedicalCollege,ZhengzhouUniversity,Zhengzhou450001,China)
Drug-induced liver injury(DILI) is a significant reason of acute liver failure and is the main cause of therapeutic drugs withdrawal from the market. Multiple mechanisms can culminate in DILI, but metabolism and genetics play distinct roles in this process. This review will cover papers we consider have addressed these mechanisms of DILI in commonly used medications for adults, and discuss the hot issues. The aim is to generate discussion about the potential clinical significance among these researchs and point out the key areas for further study of DILI.
DILI; hepatotoxicity; metabolism; genetics; HLA; CYP450
2016-01-11,
2016-04-20
国家自然科学基金资助项目(No 81173127)
王书杰(1989-),女,硕士生,研究方向:药理学,E-mail:15238617387@163.com;张莉蓉(1964-),女,博士,教授,博士生导师,研究方向:药物基因组学,通讯作者,E-mail:lrzhang@zzu.edu.cn
10.3969/j.issn.1001-1978.2016.07.003
A
1001-1978(2016)07-0898-05
R-05;R322.47;R345.99;R392.11;R575.102摘要:药物引起的肝损伤是引起急性肝功能衰竭的重要原因,也是导致治疗药物退市的主要原因。药物性肝损伤的发病机制复杂,其中以代谢、遗传学机制为主。该文综述了成人常用药物引起肝损伤的机制,并就热点问题进行讨论。旨在探讨这些研究可能的临床意义,为进一步阐明药物性肝损伤的发病机制指明方向。
网络出版时间:2016-6-20 11:49网络出版地址:http://www.cnki.net/kcms/detail/34.1086.R.20160620.1149.006.html