密度泛函理论研究CO、CO+H在Ni(111)表面的吸附
2016-07-13桂岚岚彭导灵顾凤龙
桂岚岚, 彭 亮, 彭导灵, 顾凤龙
(华南师范大学化学与环境学院,环境理论化学教育部重点实验室,广州 510006)
密度泛函理论研究CO、CO+H在Ni(111)表面的吸附
桂岚岚, 彭亮, 彭导灵, 顾凤龙*
(华南师范大学化学与环境学院,环境理论化学教育部重点实验室,广州 510006)
摘要:采用密度泛函理论(DFT)研究了CO以及CO+H体系在金属Ni(111)表面的吸附行为.采用二维平板周期性结构模型来模拟金属Ni(111)表面,消除了团簇结构模型不能考虑体系边界效应的影响,更接近于真实金属表面. 对CO在Ni(111)表面的吸附过程进行探究,结果表明:CO在不同的表面活性位吸附后C—O键不同程度被削弱;通过对吸附能以及吸附后C—O键长和C—O伸缩振动频率分析,发现顶位(top)、桥位(bridge)、六方密堆积三重穴位(hcp)和面心立方三重穴位(fcc)都以C端靠近表面的垂直吸附为稳定状态,均为非解离吸附,其中fcc与hcp两空穴位吸附性质几乎相同,为CO的最佳活性位. 研究CO与氢(H)在Ni(111)表面的吸附过程的结果表明:部分CO通过双基端加H生成中间物种—OCH和—COH, C—O键很大程度被削弱,较不加H吸附时的C—O键更容易在活性位断裂,因而以金属Ni(111)表面做催化剂的情况下H的加入有助于CO的解离.
关键词:密度泛函; Ni(111); CO+H; 费托合成; 吸附
CO是具有三重键的异核双原子分子,其中最高占据分子轨道(HOMO)轨道主要由C原子的孤对电子占据,表现出较强的配位能力.金属镍作为一种3d过渡金属具有3d84s2的电子结构,特殊的d壳层结构使其具有丰富的物理和化学性质.1902年首次发现CO和H2在镍的催化作用下可以生成甲烷,合成气甲烷化技术[1-4].目前,镍基催化剂在合成气甲烷化反应中应用前景大[5-7].纯镍晶体具有面心立方结构,其(111)面是1个原子密排面,一般情况下不发生再构,开展研究也相对简单,被广泛关注[8-17].
尽管有很多实验方法提出CO加H2在 Ni表面的催化活化机理,但受实验条件的限制尚未达成共识.ARAKI和PONEC[15]从实验中得出甲烷化反应始于CO在多核活性位上的解离化学吸附,金属表面与C原子成键,C—O键断裂,即生成金属表面Cs和O(a),CO的这种解离化学吸附快速,无需帮助,甲烷化的速率决定步骤是表面Cs连续加氢过程中的某一步;MORI等[16]发现Ni催化剂上甲烷生成速率并非取决于Cs或CHx(a)加氢的速率,并被C≡O键断裂控制,无论是氢助还是非氢助的断裂;MITCHELL等研究[17]表明,Ni(111)表面CO的分子振动频率与H原子的吸附关系不大,在混合吸附情况下,CO分子与 H原子之间的作用不强,在一定意义上,可将CO分子和H原子分开考虑,即看成2个单吸附系统的叠加.因此弄清楚Ni催化剂上CO加氢反应的主要途径,反应中CO是否解离?CO是直接解离还是加氢解离显得十分必要.基于此,本文运用量子化学计算方法研究CO加氢在金属Ni(111)表面的催化活化机理.
1计算模型和方法
纯镍在常温下是面心立方晶格(fcc),晶胞参数a=b=c=0.352 nm,α=β=γ=90°, Ni—Ni键长0.248 9 nm.本文模拟Ni(111)表面(图1中阴影部分)周期性超胞,图2为Ni(111)2×2×2的双层薄片(slab)模型中的1个超胞及其4种吸附活性,分别为顶位(top)、桥位(bridge)、六方密堆积三重穴位(hcp)、面心立方三重穴位(fcc).

图1 阴影部分表示Ni (111)表面
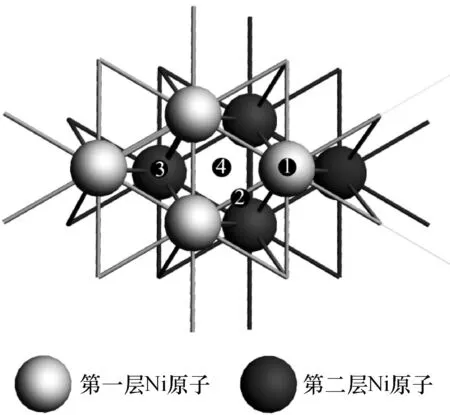
图2 Ni(111)2×2×2超胞及4种吸附活性位
Figure 2Ni (111) 2×2×2 super cell and four active adsorption sites
注:1-顶位;2-桥位;3-hcp位;4-fcc位.
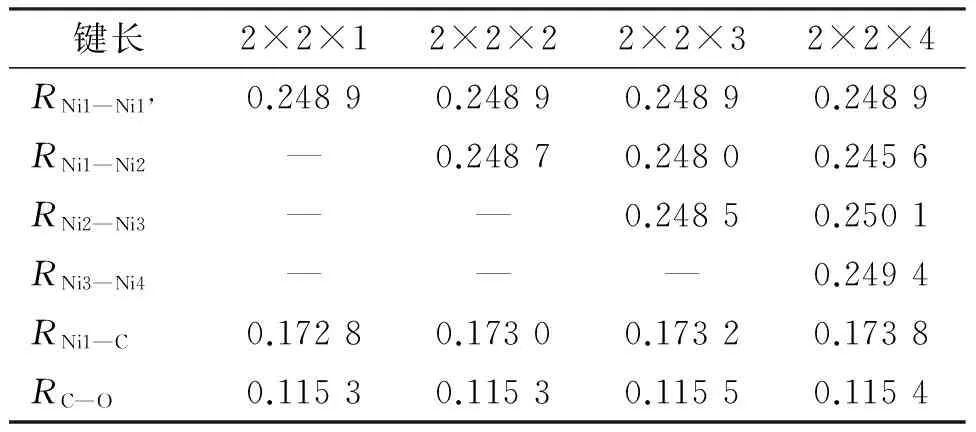
采用密度泛函理论(Density Functional Theory,DFT),选择广义梯度近似(Generalized Gradient Approximation,GGA),利用ADF(Amsterdam Density Functional)程序中能带结构模块(BAND)来计算模拟CO在具有周期性结构的Ni(111)表面的吸附过程.首先对体系的结构模型进行测试,选用CO顶位(top)吸附在Ni(111)的2×2表面,对层数设计从1递增加至4层结构体系,方法上采用交换关联相互作用GGA-PW91,基组选择芯层双zeta,价层三zeta的双极化 (TZ2P)基组,冻芯部分选用程序中的small参数,且不考虑镍元素的相对论相应,所有的收敛标准均采用程序默认标准.通过计算发现层数对Ni—Ni键长的影响几乎可以忽略(表1).本文选用了Ni(111)的2×2×2表面模型进行计算.
本文测试了3种常用的密度泛函方法B3LYP,PBE和PW91,分别让其在3种不同基组DZ、TZP、TZ2P情况下选择冻芯large或者small进行组合,测试结果见表2,数据显示采用PW91方法和TZ2P基组的计算结果与其他方法相比更接近实验值,在冻芯small时得到同一层Ni—Ni键长是0.248 9 nm,层间相邻Ni—Ni键长是0.248 7 nm,层间距离为0.219 4 nm,C—O键长0.113 6 nm;冻芯large时得到同一层Ni—Ni键长是0.248 9 nm,层间相邻Ni—Ni键长是0.248 5 nm,层间距离为0.219 4 nm,C—O键长0.113 6 nm,而同层和层间相邻Ni—Ni键长实验值都为0.248 9 nm,C—O键长实验值为0.112 8 nm.不同冻芯参数的选择对结果影响很小,所以本文选用PW91方法TZ2P基组,以及冻芯为small来节省计算时间.
表11-4相邻层Ni—Ni、Ni—C及C—O键长
Table 1Bond lengths of Ni—Ni, Ni—C and C—O bonds in different layer (1-4) models
nm
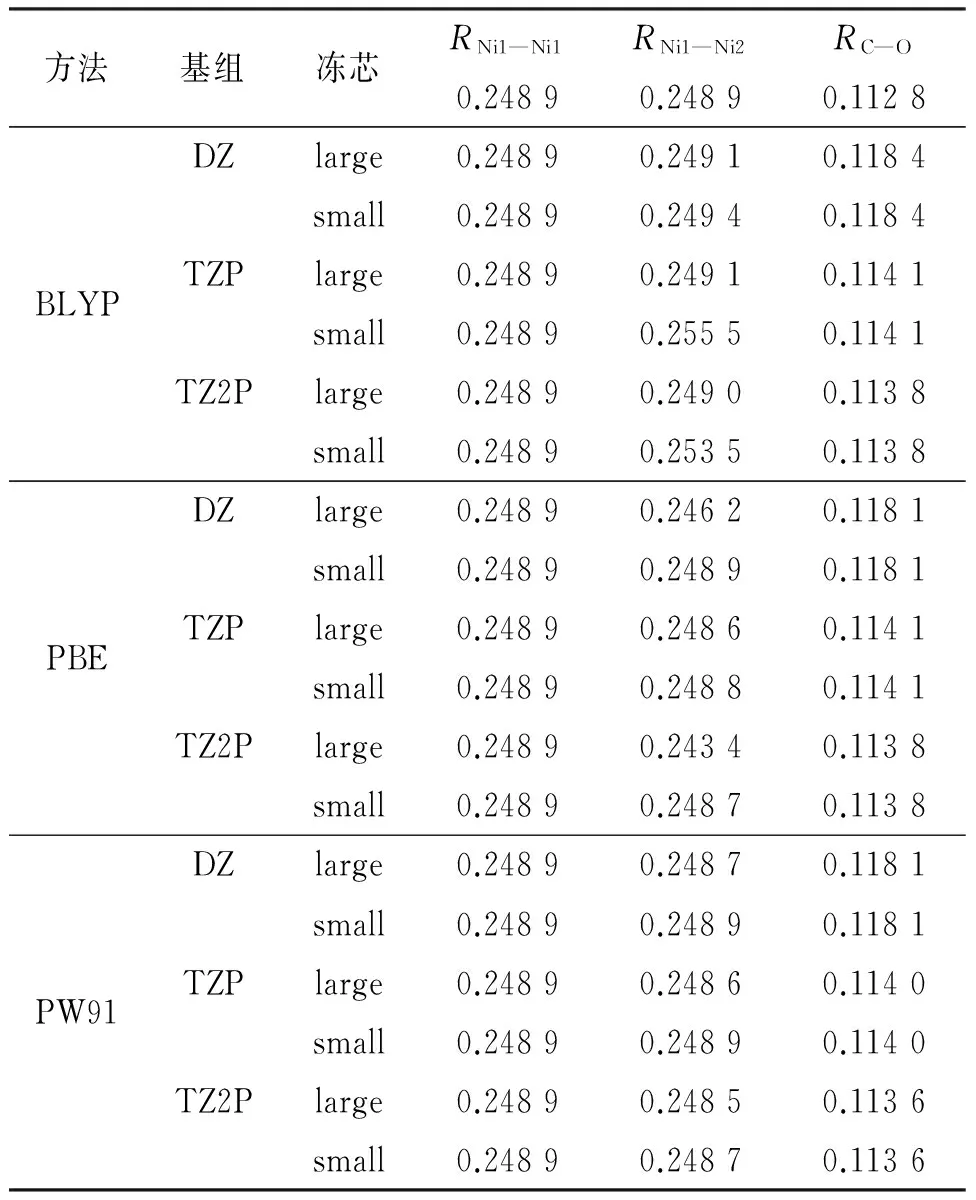
表2 BLYP、PBE及PW91方法与基组的组合测试
2结果与讨论
2.1CO在Ni(111)表面的吸附
分别对CO在Ni(111) 表面上的顶位(top)、桥位(bridge)、六方密堆积三重穴位(hcp)、面心立方三重穴位(fcc)4种活性位的C端接近垂直吸附,O端接近垂直吸附与平躺吸附的12种不同初始结构进行几何优化(图3),图中标记(1)、(2)分别表示表面结构的平视图与俯视图.
经过对初始的12种结构进行优化,得到CO分子在Ni(111) 表面的4种不同吸附活性位置的稳定结构(图4),从成键角度分析,top位吸附稳定状态键角O—C—Ni1为180°,则C、O与Ni(111)表面Ni1原子恰成直线,且O、C、Ni1原子(x,y)坐标相同,可见CO垂直吸附于Ni1顶位(top);bridge位吸附稳定状态中C、O原子(x,y)坐标相同,键角O—C—Ni1与角O—C—Ni3相等(139.1°),则CO恰为垂直吸附于bridge位活性位;而hcp位、fcc位2种吸附状态非常相似,C、O两原子(x,y)坐标仍然相同,且与之形成相应穴位的表面的3个Ni原子形成的三键角相等,六方密堆积三重穴位(hcp)中三键角O—C—Ni3、O—C—Ni5、O—C—Ni7均为132.9°,fcc位中三键角O—C—Ni1、O—C—Ni3、O—C—Ni5均为132.6°,即CO同样是垂直吸附于hcp和fcc位.综上所述,4种稳定状态均是CO以碳原子接近Ni(111)表面的垂直吸附(图4).

图3 4种活性位置12种初始结构
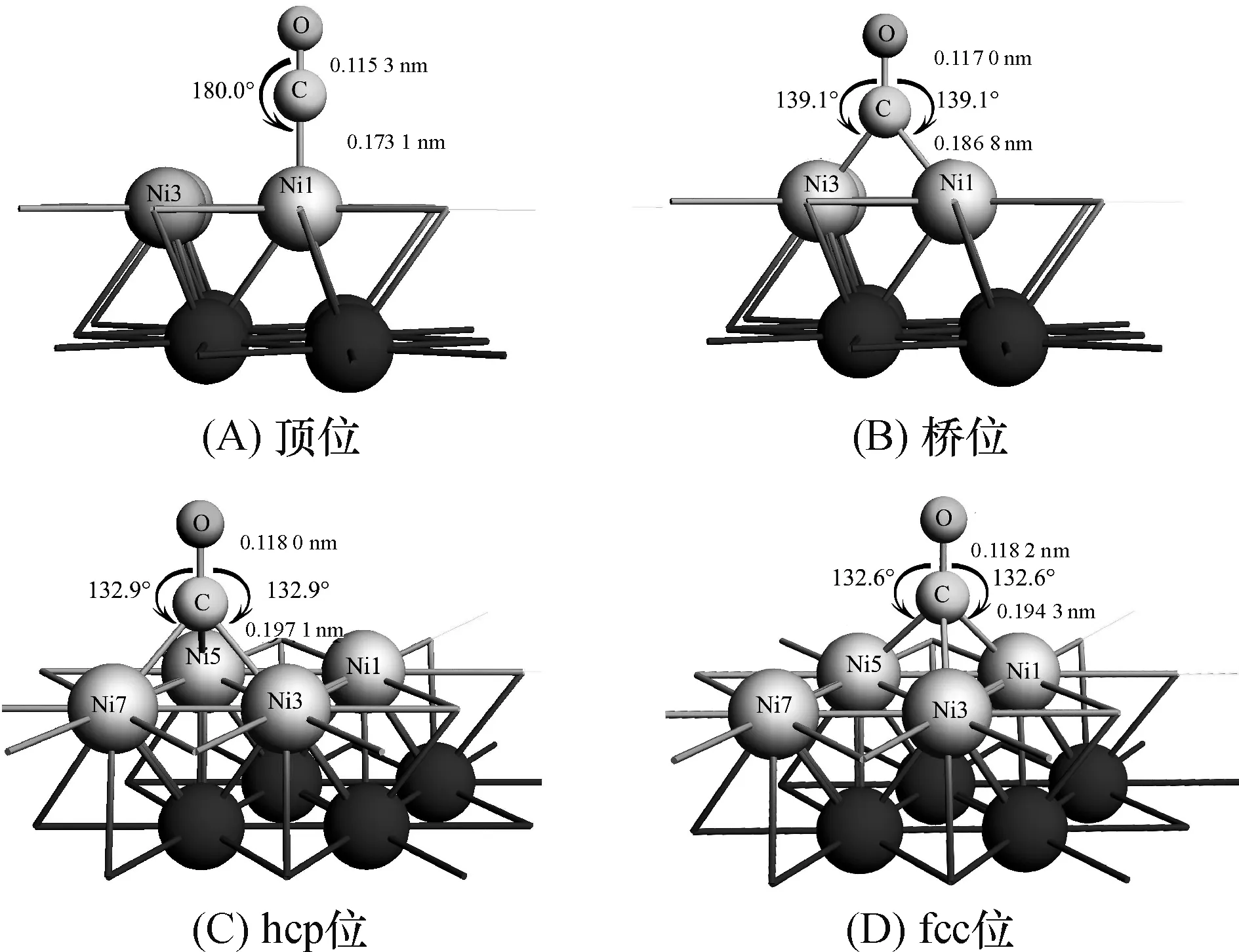
图4 4种吸附活性位的稳定结构
表3显示CO分子在Ni(111) 表面吸附时的吸附能、键长和吸附后C—O振动频率计算值.吸附能是指吸附后体系的总能量与未产生吸附的洁净金属表面和CO自由分子的能量之差Eads=E(surface+CO)-E(surface)-E(CO).吸附能为负值表明吸附过程为自发,其绝对值越大,则吸附结构越稳定,吸附位的活化性能越高.从表3看出,4种吸附结构的吸附能值接近,表明CO在Ni(111)金属表面的扩散能量壁垒很低,尤其是bridge位、hcp位、fcc位3种结构吸附能接近,但同时,hcp位和fcc位2种吸附体系无论是吸附能、Ni—C键长、C—O键长与C—O振动频率大小都非常接近,为2种极其相似的稳定吸附状态.气体CO分子中C—O键长计算值为0.113 6 nm,与实验值0.112 8 nm相差仅为1.1%,CO在镍金属表面top位吸附后C—Ni键长为0.173 1 nm,在bridge位吸附后C—Ni 键长为0.186 8 nm,hcp位吸附后C—Ni键长为0.194 1 nm,fcc位吸附后C—Ni键长为0.194 3 nm,Ni—C键的计算结果与Ni—CO原子簇模型采用UHF/STO-3G方法[18-19]的计算值(0.184 0)、HF/大基组方法[20]的计算值(0.207 0)以及GVB(2/4)/DZ方法[21]的计算值(0.194 0)等结果基本吻合(表3).

表3 4种稳定结构吸附能、Ni—C和C—O键长以及C—O频率
从表3看出,hcp位和fcc位吸附时,C—O键长分别为0.118 0 nm和0.118 2 nm,较顶位(0.115 3 nm)和桥位(0.117 0 nm)分别高出0.003 nm和0.001 nm,表明hcp位和fcc位的吸附位活化性能比顶位和桥位高.同时通过吸附前后C—O振动频率分析可知,气相CO的振动频率为2 179 cm-1,这比实验值2 143 cm-1略大.计算结果表明,吸附后CO的振动频率与气相值相比均发生不同程度的红移,且结构越稳定红移程度越大,这与吸附后C—O键长的增加相一致.fcc位C—O的振动频率(1 821 cm-1)最小,从而进一步证实了fcc位的活化CO分子的性能最好.
对于活化性能最好的fcc位,为寻找其可能的过渡态结构,采用固定C—O键长分别为0.11、0.12和0.13 nm,Ni—C键长从0.175 nm到0.500 nm的变化过程作一系列的单点,计算得到曲线如图5所示.随着Ni—C键长增至0.300 nm以后,各结构的单点能保持平稳,说明fcc活性位置吸附无过渡态,只是单纯的吸附过程.
对CO与Ni(111)表面的成键过程进行态密度(DOS)分析(图6),其中发挥作用的主要是5σ和2π*轨道,形成了 5σ成键和Ni-2π*反键.CO的5σ轨道上的电子转移至Ni的d轨道,使5σ轨道发生左移,Ni的d轨道上的电子转移到CO的2π*轨道,使2π*轨道发生分裂,部分进入成键轨道.
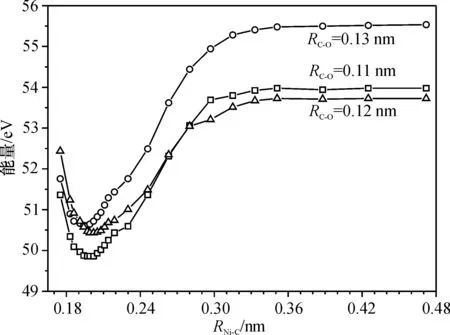
图5在fcc吸附位固定C—O链长时不同Ni—C键长单点能曲线
Figure 5Energies of different Ni-C bonds in fcc adsorption site with the fixed C—O bond lengths
2.2CO+H在Ni(111)表面的吸附
在本节中初始结构设计基于上一节CO单吸附于Ni(111) 表面上的顶位、桥位、hcp位、fcc位的以C端接近垂直吸附,O端接近垂直吸附,与平躺吸附的12种位置上C端或O端各加上1个H,图7显示,顶位的6种构造,4种活性位置共构造出24种不同的初始结构.
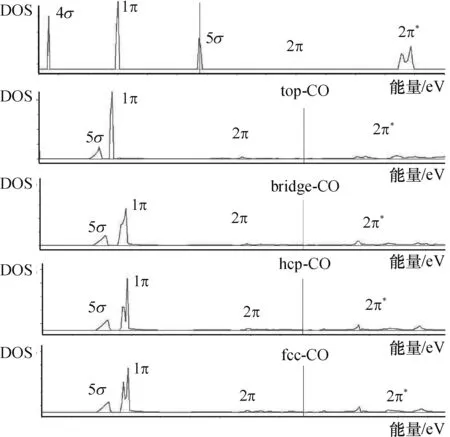
图6 CO在Ni(111)表面的分波态密度
优化24种初始结构得到CO+H在Ni(111) 表面的8种不同稳定结构(图8).其中Ⅳ、Ⅴ、Ⅶ、Ⅷ 4种结构有相同的特点,CO分子与H之间不成键,而是相互远离地吸附在Ni(111)表面的三重活性穴位fcc或者hcp上.结构Ⅳ中CO吸附于fcc位,同时,H吸附于其对顶的hcp位上;结构Ⅴ中CO吸附于fcc
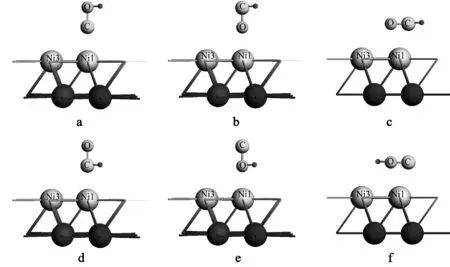
图7 CO+H在Ni(111) 表面顶位的6种初始结构
Figure 7Initial structures of CO+H on Ni (111) surface at top site
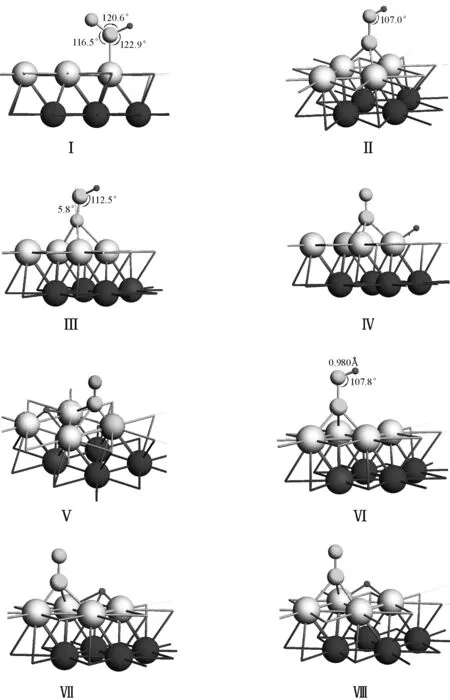
图8 CO+H在Ni(111) 表面的8种稳定结构
位,而H吸附于其同单元相邻的hcp位上;结构Ⅶ中CO吸附于hcp位,而H吸附于其同单元相邻的fcc位上;结构Ⅷ中CO与H分别处在相间的hcp位上.这4种稳定结构不但能量相当,其C—O键、Ni—C键、Ni—H键以及C—O振动频率与CO和H分别单吸附在Ni(111)表面相同活性位的吸附构型几乎保持一致(表4),研究得出的CO分子与 H原
表48种稳定态能、C—O键长、Ni—C距离、Ni—H距离、C—O频率及CHO夹角
Table 4Stabilization energies, C—O bond lengths, distances of Ni—C, distances of Ni—H, vibrational frequencies of C—O and angles of CHO in different stable structures
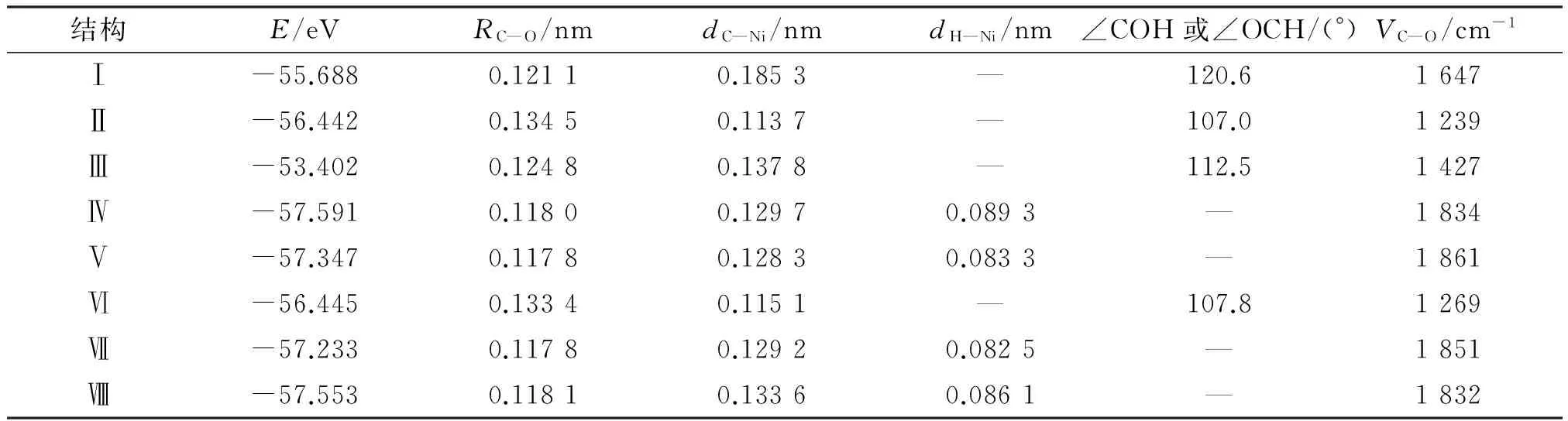
结构E/eVRC—O/nmdC—Ni/nmdH—Ni/nm∠COH或∠OCH/(°)VC—O/cm-1Ⅰ-55.6880.12110.1853—120.61647Ⅱ-56.4420.13450.1137—107.01239Ⅲ-53.4020.12480.1378—112.51427Ⅳ-57.5910.11800.12970.0893—1834Ⅴ-57.3470.11780.12830.0833—1861Ⅵ-56.4450.13340.1151—107.81269Ⅶ-57.2330.11780.12920.0825—1851Ⅷ-57.5530.11810.13360.0861—1832
子之间的作用不强,这与MITCHELL等[17]176提出的将共吸附系统看成2个单吸附系统叠加的结论一致.
结构Ⅰ为以碳原子一端靠近Ni 原子顶位的垂直吸附,O原子和H原子分别位于C原子的两边成一定的夹角,与CO单吸附在Ni(111)表面时的顶位吸附稳定结构比较, C—O键长由单吸附时0.115 3 nm增长到0.121 1 nm,C—O伸缩振动频率由单吸附的2 070 cm-1降到1 647 cm-1,显然C—O键有所削弱,此时的C、H、O 3原子与活性位Ni原子4点共面,这也是计算结果中唯一的以顶位为活性位的稳定结构;结构Ⅱ与Ⅵ相似,前者活性位为fcc位,后者活性位为hcp位,分别与CO单吸附在Ni(111)表面上的fcc位与hcp位的稳定结构相比较,都以C端垂直吸附在各自活性位上,不同之处在于,在H与O形成O—H键的同时C—O键明显增长,fcc位由之前的0.118 2 nm增长到0.134 5 nm,C—O伸缩振动频率由单吸附的1 821 cm-1降到1 239 cm-1,hcp位由之前的0.118 0 nm增长到0.133 4 nm,C—O伸缩振动频率由单吸附的1 833 cm-1降到1 269 cm-1,这使C—O键受到了较大程度的削弱.
结构Ⅲ是计算结果中唯一的以O端靠近活性位的稳定结构,C—O键长(0.124 8 nm)与CO单吸附时的C—O键一样有所削弱,方向上与Ni表面垂直方向偏差5.8°,而在CO单吸附于Ni(111)表面的4种稳定结构中均为C端垂直吸附,由此说明,CO在Ni(111)表面上的催化活性在加氢前后有较大差异,表现为加氢有助于C—O键的削弱,可以推断—OCH或—COH断裂为—CH和—O或—OH和—C的过程比在相同Ni(111)表面活性位上CO分子断裂为—C和—O的过程容易.
3结论
采用密度泛函理论研究CO,CO+H在金属Ni(111)-2×2×2表面催化反应中C—O键的活化机理,根据计算结果得出以下结论:
(1)CO单吸附于Ni(111) -2×2×2表面体系中,CO均以C端靠近且垂直吸附于Ni(111)表面的顶位(top)、桥位(bridge)、六方密堆积三重穴位(hcp)、面心立方三重穴位(fcc)4种活性位上,C—O键各有不同程度的削弱,且削弱强度top (2)CO加H吸附于Ni(111) -2×2×2表面体系中,一是CO和H直接被Ni(111)表面的活性位hcp或fcc吸附,CO和H之间相邻、对顶或相间但互不成键,可视为CO与H各自单吸附于Ni(111)同活性位系统的叠加;二是H原子通过与CO分子的双基端C或O成键生成中间物种—OCH和—COH,此时C—O键已成为单键,与CO单吸附于Ni(111)过程相比很大程度被削弱.综上所述,H的加入有助于C—O键在Ni(111)表面的活化. 参考文献: [1]RABOU L P, BOS L. High efficiency production of substitute natural gas from bio-mass[J]. Applied Catalysis B:Environmental, 2012,111(2):456-460. [2]GRÖBL T, WALTER H, HAIDER M. Biomass steam gasification for production of SNG-process design and sensitivity analysis[J]. Applied Energy, 2012,97(3):451-461. [3]MEIJDAN C M V D, VERINGA H J, RABOU L P M. The production of synthetic natural gas(SNG):a comparison of three wood gasification systems for energy balance and overall efficiency[J]. Biomass & Bioenergy, 2010,34(3):302-311. [4]SCHILDHAUER T J, SEEMANN M C, BIOLLAZ S M A. Fluidized bed methanation of wood-derived producer gas for the production of synthetic natural gas[J]. Industrial & Engineering Chemistry Research, 2010,49(15):7034-7038. [5]KUSTOV A L, FREY A M, LARSEN K E, et al. CO methanation over supported bimetallic Ni-Fe catalysts:from computational studies towards catalyst optimization[J].Applied Catalysis A General, 2007,320:98-104. [6]PANAGIOTOPOULOU P, KONDARIDES D I, VERYKIOS X E. Selective methanation of CO over supported noble metal catalysts:effects of the nature of the metallic phase on catalytic performance[J]. Applied Catalysis A: General, 2008,344(1):45-54. [7]TAKENAKA S, SHIMIZU T, OTSUKA K. Complete removal of carbon monoxide in hydrogen-rich gas stream through methanation over supported metal catalysts[J]. International Journal of Hydrogen Energy, 2004,29(10):1065-1073. [8]MORIKAWA Y, MORTENSEN J J, HAMMER B, et al. CO adsorption and dissociation on Pt(111) and Ni(111) surfaces[J]. Surface Science, 1997,386(1):67-72. [9]DAVIS R, WOODRUFF D P, HOFMANN P, et al. Local structure determination for low-coverage CO on Ni(111)[J]. Journal of Physics: Condensed Matter, 1996,8(10):1367-1379. [10]ERLEY W, WAGNER H, IBACH H. Adsorption sites and long range order-vibrational spectra for CO on Ni(111)[J]. Surface Science, 1979,80(79):612-619. [11]SCHAFF O, FERNANDEZ V, HOFMANN P, et al. Coverage-dependent changes in the adsorption geometry of benzene on Ni{111}[J]. Surface Science, 1996,348(95): 89-99. [12]FERNANDEZ V, SCHINDLER K M, SCHAFF O, et al. Structure determination of a CO/O coadsorption phase on Ni(111)[J]. Surface Science, 1996,351(1): 1-12. [13]BECKER L, AMINPIROOZ S, HILLERT B, et al. Threefold-coordinated hollow adsorption site for Ni(111)- c(4×2)-CO: a surface-extended X-ray-absorption fine-structure study[J]. Physical Review B: Condensed Matter, 1993,47(15):9710-9714. [14]CAMPUZANO J C, GREENLER R G. The adsorption sites of CO on Ni (111) as determined by infrared reflection-absorption spectroscopy[J]. Surface Science, 1979,83(1):301-312. [15]ARAKI M, PONEC V. Methanation of carbon monoxide on nickel and nickel-copper alloys[J]. Catalysis, 1976, 44(3): 439-448. [16]MORI T, MASUDA H, IMAI H, et al. Kinetics, isotope effects, and mechanism for the hydrogenation of car bon monoxide on supported nickel catalysts[J]. Physical Chemistry, 1982,86(14): 2753-2760 [17]MITCHELL G E, GLAND J L, WHITE J M. Vibrational spectra of coadsorbed CO and H on Ni(100) and Ni(111)[J]. Surface Science, 1983,131(1):167-178. [18]XU X, WANG N Q, ZHANG Q. Chemisorption on metal surfaces: cluster model studies[J].Bulletin of the Chemical Society of Japan, 1996,69(3):529-534. [19]XU X, WANG N Q, ZHANG Q. Chemisorption on metal surfaces: cluster model studies[J]. Surface Science, 1992,274(3):378-385. [20]GODDARD W A, WALCH S P, RAPPE A K, et al. Methanation of CO over Ni catalyst: a theoretical study[J]. Vacuum Science and Technology, 1977,14(1):416-418. [21]AVOURIS P, BAGUS P S, ROSSI A R. Excitation and ionization at surfaces: CO on metals[J]. Journal of Vacuum Science & Technology B: Microelectronics Processing and Phenomena, 1985,3(5):1484-1489. [22]BERTOLINI J C, DALMAI-IMELIK G, ROUSSEAU J. Benzene adsorption on nickel (100) and (111) faces studied by LEED and high resolution electron energy loss spectroscopy[J]. Surface Science, 1977,67(2):478-480. [23]DAVILA M E, ASENSIO M C, WOODRUFF D P, et al. Structure determination of Ni(111)c(4×2)-CO and its implications for the interpretation of vibrational spectroscopic data[J]. Surface Science, 1994,311(3):337-348. [24]STEININGER H, LEHWALD S, IBACH H. On the adsorption of CO on Pt(111)[J]. Surface Science, 1982,123(2):264-282. [25]SURMEV L, XU Z, YATES J T. IRAS study of the adsorption of CO on Ni(111): interrelation between various bonding modes of chemisorbed CO[J]. Surface Science, 1988,201(1):1-13. 【中文责编:谭春林英文责编:李海航】 Density Functional Theory Study of CO, CO+H Adsorption on Ni(111) Surface GUI Lanlan, PENG Liang, PENG Daoling, GU Fenglong* (School of Chemistry and Environment, South China Normal University,Key Laboratory of Theoretical Chemistry of Environment, Ministry of Education, Guangzhou 510006, China) Abstract:A density functional theory (DFT) study of CO, CO+H adsorption processes on Ni(111) surface is reported in this article. A two-dimensional periodic slab structure model is employed to simulate the Ni (111) surface and to eliminate the influence of the cluster model on system boundary effects, making the model closer to the true metal surface. This article is divided into two parts. The adsorption process of CO on Ni (111) surface is studied first, and the results showed that the C—O bonds are weakened in different surface active adsorption sites. According to the analysis of adsorption energies, C—O bond lengths and the C—O stretching vibration frequencies, it is found that there exist 4 adsorption sites which are the top, bridge, hexagonal close packing (hcp), and face centered cubic (fcc) sites. C—O bond is perpendicular to the metal surface and the C atom is in the near-metal position in the stable state of all adsorption sites. All of the stable states are non-dissociative adsorption states. The properties of CO adsorbed on the fcc and hcp sites are almost the same. The adsorption process of CO and H on Ni (111) surface is studied next, and the results showed that CO combined with H to generate the intermediate species —OCH and —COH. By analyzing the C—O bond length and the stretching vibration frequency, it is found that the C—O bond is largely weakened. The C—O bond can be broken down more easily in the active site comparing with CO adsorption without H. Therefore, with metal Ni (111) surface as catalyst, the added hydrogen is helpful for the dissociation of CO. Key words:density functional theory; Ni(111); CO+H; Fischer-Tropsch synthesis; adsorption 收稿日期:2015-03-08《华南师范大学学报(自然科学版)》网址:http://journal.scnu.edu.cn/n 基金项目:国家自然科学基金项目(21273081) *通讯作者:顾凤龙,教授,Email:gu@scnu.edu.cn. 中图分类号:O621 文献标志码:A 文章编号:1000-5463(2016)01-0067-07
