大肠杆菌磷酸甘油酸脱氢酶突变体的构建及抗反馈抑制效应
2016-07-04邓辉陈存武孙传伯韦传宝
邓辉,陈存武,孙传伯,韦传宝
1 皖西学院 生物与制药工程学院,安徽 六安 237012 2 六安市蛋白质分离与纯化研究中心,安徽 六安 237012
大肠杆菌磷酸甘油酸脱氢酶突变体的构建及抗反馈抑制效应
邓辉1,2,陈存武1,孙传伯1,韦传宝1,2
1 皖西学院 生物与制药工程学院,安徽 六安237012 2 六安市蛋白质分离与纯化研究中心,安徽 六安237012
邓辉, 陈存武, 孙传伯, 等. 大肠杆菌磷酸甘油酸脱氢酶突变体的构建及抗反馈抑制效应. 生物工程学报, 2016, 32(4): 468–477.
Deng H, Chen CW, Sun CB, et al. Construction and characterization of Escherichia coli D-3-phosphoglycerate dehydrogenase mutants with feedback-inhibition relief. Chin J Biotech, 2016, 32(4): 468–477.
摘要:磷酸甘油酸脱氢酶 (D-3-phosphoglycerate dehydrogenase,PGDH,EC 1.1.1.95) 为L-丝氨酸合成途径的关键酶,其编码基因为serA,其活性受到合成产物L-丝氨酸的反馈抑制调控。为解除丝氨酸的反馈抑制,采用定点突变技术把编码PGDH酶344位组氨酸或346位天冬氨酸或364位天冬氨酸的密码子定点突变为丙氨酸密码子。改造后的serAFbr被连到表达载体pT7-7上,并转入大肠杆菌Escherichia coli BL21 (DE3) 中进行表达,破壁回收粗酶液,通过DEAE阴离子柱纯化PGDH突变体,并对其酶活性和IC50值进行了测定。结果,野生型PGDH酶IC50值为7 μmol/L,而PGDH双突变体N346A/H344A催化活性与野生型相近,在丝氨酸浓度为160 mmol/L时,其酶活仍保持未添加丝氨酸时酶活的96%,基本解除反馈抑制。
关键词:大肠杆菌磷酸甘油酸脱氢酶,突变体的构建,解除反馈抑制
Received: July 27, 2015; Accepted: October 29, 2015
Supported by: National High Technology Research and Development Program (863 Program) (No. 2012AA021500), National Basic Research Program of China (973 Program) (No. 2012CB720805), National Natural Science Foundation of China (Nos. 81274021, H2801).
Hui Deng. Tel/Fax: +86-564-3305073; E-mail: dhup@qq.com
国家高技术研究发展计划 (863计划) (No. 2012AA021500),国家重点基础研究发展计划 (973计划) (No. 2012CB720805),国家自然科学基金 (Nos. 81274021,H2801) 资助。
L-丝氨酸处于氨基酸代谢的中间位置,参与许多生物物质的合成,具有许多重要的生理功能和作用,食品和医药产业对其需求日益增大[1]。磷酸甘油酸脱氢酶 (PGDH,d-3-phosphoglycerate dehydrogenase,EC 1.1.1.95) 为L-丝氨酸合成途径的关键酶,催化L-丝氨酸生物合成的第一步,其编码基因为serA,其活性受到合成产物L-丝氨酸的反馈抑制调控[2],它的克隆表达及反馈抑制的解除将有助于构建在生产上高产L-丝氨酸及其代谢衍生物的生产菌株。
David等[3]在0.9 Å分辨率下解析了结合有丝氨酸的磷酸甘油酸脱氢酶晶体结构,晶体结构分析表明:磷酸甘油酸脱氢酶为四聚体,每个亚基由核苷酸结合域、底物结合域和调控域组成。亚基之间的相互作用发生在调控区域,该区域形成一延伸的β折叠结构,产物L-丝氨酸的结合位点就位于这个表面。晶体结构给出了与抑制物丝氨酸成极性共价键的氨基酸残基,变构调控结合位点见图1。
为解除合成产物L-丝氨酸对磷酸甘油酸脱氢酶的反馈抑制调控,本研究直接阻断丝氨酸与酶的极性共价结合,即利用定点突变技术把磷酸甘油酸脱氢酶上与丝氨酸结合的344位组氨酸或346位天冬氨酸或364位天冬氨酸突变为丙氨酸,使丝氨酸不能与PDGH形成极性共价键,致使丝氨酸无法完成对PDGH的变构调控,从而获得基本解除丝氨酸对PDGH反馈抑制作用的突变体。

图1 丝氨酸分子在PGDH酶上结合位点Fig. 1 Serine bonding sites of PGDH. Subunit interface of PGDH consisting of two regulatory domains is shown in cartoon form. Banding subunit A is depicted in green, banding subunit D in purple. The two allosteric serine ligands are drawn in light blue, and side chains of residues which form important contacts (yellow dotted line) to the ligands are especially shown in molecular structural form. Using PyMOL 1.4 software to simulate the above picture.
1 材料与方法
1.1材料
1.1.1菌株、载体和质粒
菌株BW25113、JM109和BL21 (DE3),质粒pMDTM18-T Simple均购自Sigma公司。质粒pT7-7为本实验室保存。质粒pMDTM18-T-serA、pMDTM18-T-serAFbr、pT7-7-serA和pT7-7-serAFbr为本次试验构建,其中Fbr为feedback-inhibition resistance缩写,即抗反馈抑制。
1.1.2培养基、工具酶和试剂
培养基配方见文献[4],NdeⅠ、Hind Ⅲ、DpnⅠ、CIAP及T4 DNA连接酶购自TaKaRa 公司;pMDTM18-T Simple、基因组DNA 及质粒小量提取试剂盒为Promega 公司产品;蛋白定量试剂盒与酶活性测定试剂为Sigma 公司产品;酵母提取物和胰蛋白胨为Oxoid 公司产品;其他试剂为国产分析纯。
1.1.3PCR引物设计
根据GenBank 中报道的E. coli str.K-12 Substr. MG1655的serA 基因序列 (Accession No. 945258),以构建PGDH双突变体N346A/ H344A为例,设计4条寡核苷酸引物,并在两端加上合适的酶切位点 (NdeⅠ,Hind Ⅲ) (表1),引物合成及测序由上海生工生物技术有限公司完成。

表1 本研究所用的引物名称与序列Table 1 Primers used in this study
1.2重组表达质粒的构建
用基因组提取试剂盒提取E. coli K-12 BW25113基因组DNA,用引物系列1 PCR扩增得到的野生型serA基因,将其与克隆载体pMDTM18-T Simple连接后,转入JM109进行扩增,借助重组克隆质粒pMDTM18-T-serA,用引物系列2对serA进行H344A、N346A定点突变,获得重组克隆质粒pMDTM18-serAFbr,然后把野生型serA基因、突变型serAFbr基因和pT7-7载体用NdeⅠ、Hind Ⅲ双酶切后分别切胶回收,用T4 DNA连接酶进行连接,获得重组表达质粒pT7-7-serA和pT7-7-serAFbr。
1.3野生型及突变型酶基因的表达和产物的纯化
把重组表达质粒pT7-7-serA和pT7-7-serAFbr电转入BL21 (DE3),取活化后菌体100 μL菌液至50 mL TB培养基,37 ℃培养至OD600约为1.3时,转入25 ℃摇床继续培养0.5 h,添加IPTG至终浓度50 mmol/L[5],继续培养10−16 h。
取100 mL培养后的菌体12 000 r/min、4 ℃离心,去尽上清,加入10 mL PBS缓冲液(20 mmol/L 磷酸钾缓冲液,1 mmol/L DTT,pH 7.5),充分混匀,ON 10 s,OFF 1 min超声破碎,使浑浊液澄清。4 ℃、13 000 r/min离心30 min,上清和沉淀分别–20 ℃保藏。
用AKTA蛋白纯化系统,采用Sepharose Fast Flow层析柱,DEAE为其阴离子交换介质基团,洗脱液为:A液 (20 mmol/L 磷酸钾缓冲液,1 mmol/L DTT),B液 (20 mmol/L磷酸钾缓冲液,1 mmol/L DTT,1 mol/L NaCl)。对出峰蛋白进行酶活测定,选取合适的洗脱液比例洗脱目的蛋白。用30 kDa的纤维柱浓缩蛋白样液。
用SDS-PAGE蛋白电泳分析纯化产物和粗酶液,比较纯化效果。
1.4酶活性测定
PGDH酶活性的测定见文献[6]。2 mL反应体系中含有:40 mmol/L磷酸钾缓冲液 (pH 7.5),1.0 mmol/L DTT,0.25 mmol/L NADH,5 mmol/L α-KG,10−20 μL 的已纯化蛋白。测定方法:把0 ℃酶液加入37 ℃的反应体系,测定样品在340 nm条件下的NADH的吸光值减少量,计算酶的活性。1 U酶活定义等于反应体系中每分钟减少1 nmol/L NADH。蛋白浓度测定采用Bradford法[7],绘出蛋白浓度标准曲线,根据酶活和蛋白含量计算比酶活。
1.5野生型及突变型PGDH稳态动力学性质比较及抗反馈抑制效果
测定不同浓度α-KG (α-酮戊二酸) 下的反应速度,作1/[s]和1/[v]的双倒数曲线图,求出反应体系为:40 mmol/L磷酸钾缓冲液 (pH 7.5),1.0 mmol/L DTT,0.25 mmol/L NADH,不同浓度α-KG,10−20 μL的已纯化蛋白。按1.4酶活性测定方法测定酶活。
IC50表示在底物浓度饱和的情况下,抑制原酶活性50%时丝氨酸的浓度。在PGDH酶活(μmol/L) 分别为0、4、8、9、15、20、30、40、50、60、80、20 000、40 000、60 000、80 000、160 000,加入纯化后的野生型及突变型酶蛋白10 μg,以未加入丝氨酸时的酶活性为100%,计算酶活比。
1.6野生型及突变型PGDH理化性质比较
测定不同反应温度下 (30−70 ℃) 酶液的酶活来确定最适温度。反应体系见1.4酶活性测定,酶液在待测温度下先预热1 min后加入同样温度下预热2 min的反应液中,待测温度下测定样品在340 nm条件下的NADH的吸光值减少量,计算酶的活性。
测定不同温度 (0−60 ℃) 下保存3 min后的酶液的酶活来确定热稳定性。按1.4酶活性测定方法确定酶活比值。
测定不同pH值下 (pH 6−9) 酶液的酶活来确定最适pH。反应体系为:40 mmol/L磷酸钾-钠缓冲液 (pH 6−9) (由Na2HPO4和KH2PO4混合),1.0 mmol/L DTT,0.25 mmol/L NADH,5 mmol/L α-KG,10−20 μL的已纯化蛋白。酶液调整到待测pH值下加入同样pH值反应液中,按1.4酶活性测定方法确定酶活比值。
测定在磷酸钾-钠缓冲液不同pH值 (pH 6–9) 下4 ℃保存24 h后酶液的酶活来确定酸碱稳定性。按1.4酶活性测定方法确定酶活比值。
2 结果与分析
2.1野生型和突变型重组表达质粒的构建
对比L-丝氨酸结合前后的PDGH的晶体结构,可知当一个L-丝氨酸分子与PDGH一个亚基结合域的两个氨基酸残基344H、346N和相邻亚基结合域的364’N形成3个极性共价键时,就会引起两亚基间的空间构像改变,进而引起整个晶体的空间结构变化,为最大限度维持原蛋白晶体的构像,又尽量解除丝氨酸对PDGH两相邻亚基的作用,实验选择突变H344A或N346A或N364A来解除L-丝氨酸的反馈抑制。
以双突变体PDGH (N346A/H344A) 为例,用引物系列1从基因组扩增出serserA,构建重组克隆质粒pMDTM18-T-serserA,用引物系列2 对serA进行H344A、N346A定点突变,获得pMDTM18-T-serAFbr,再把pMDTM18-T-serA、pMDTM18-T-serAFbr和pT7-7[8]载体酶切后连接,获得重组表达质粒pT7-7-serA和pT7-7-serAFbr。图2为重组表达质粒pT7-7-serA及pT7-7-serAFbr的双酶切鉴定结果。

图2 重组质粒pT7-7-serA及pT7-7-serAFbr的酶切鉴定Fig. 2 Identification by restriction digestion on recombinant plasmids of pT7-7-serA and pT7-7-serAFbr. M1: DS 15 000 marker; M2: DL 2 000 DNA marker; 1: recombinant plasmids of pT7-7-serA after enzyme digestion; 2: recombinant plasmids of pT7-7-serAFbrafter enzyme digestion.
测序结果表明:所构建成重组质粒pMD18-T-serA中serA的核苷酸序列与GenBank 中报道的E. coli K-12 的serA 基因序列一致;所构建的重组质粒pMD18-T-serAFbr的H344A、N346A定点突变成功。
2.2PGDH酶液的分离和纯化
由于该酶作为 α-酮戊二酸 (α-ketoglutarate, α- K G ) 还原酶和作为3-磷酸甘油酸(3-phosphoglycerate,3PG) 脱氢酶的稳态动力学特性一致,且二者对终产物L-丝氨酸反馈抑制特性也几乎完全一致[9],以α-KG作为底物测该酶3PG脱氢酶活性的方法近年来也得到研究者的普遍应用[10-15],所以我们采用测定α-酮戊二酸 (α-ketoglutarate,α-KG) 还原酶特性代替测定3PG (3-phosphoglycerate,3PG) 脱氢酶的特性。
高速离心菌液收集诱导表达PDGH后的菌体,加入适量缓冲液混匀后超声破碎至澄清,再高速离心取上清 (即粗酶液) 于–20 ℃保藏。用DEAE阴离子柱洗脱目的蛋白,用30 kDa的纤维柱浓缩蛋白样液,用SDS-PAGE蛋白电泳对纯化产物和粗酶液进行电泳,比较纯化效果。PGDH酶及其突变体的蛋白分离纯化步骤参数见表2。
由表2知纯化后的PGDH比酶活约为1.0×104U/mg,比纯化前的比酶活提高了约3.2倍。SDS-PAGE电泳比较纯化后酶液和粗酶液,结果见下图3。
由图知纯化后的PDGH亚基蛋白分子量约在45 kDa,纯化后的目的条带清晰,达到电泳纯。
2.3野生型及突变型PGDH稳态动力学性质比较及抗反馈抑制效果
在NADH浓度饱和下,测定不同浓度α-KG下的反应速度,作1/[s]和1/[v]的双倒数曲线图,得出PDGH的α-KG还原酶动力学参数,结果见表3。

表2 不同处理步骤对PGDH纯化的影响Table 2 The influence of different processing steps on PGDH purification

图3 野生酶与突变酶SDS-PAGE蛋白电泳Fig. 3 SDS-PAGE protein electrophoresis of wild type enzyme and its mutation. 1, 2: wild type enzyme and its mutation after purification; 3, 4: soluble cellular fraction of BL21(DE3) (serA) and its mutation; M: protein marker (14.4 kDa−94.0 kDa).

表3 野生型PDGH酶及其突变体的动力学参数Table 3 Kinetic parameters of Wild type PDGH and Mutant
L-丝氨酸对突变体和野生型PDGH的α-KG还原酶活性为非竞争性抑制,在PGDH 酶活性测定反应体系中加入一系列不同浓度的L-丝氨酸,测定丝氨酸对野生型及突变型PGDH的活性影响。不同丝氨酸浓度下野生酶和突变体(N346A/H344A) 真实酶活和酶活比的变化见图4。
由曲线数据显示,未添加L-丝氨酸时,野生酶和突变酶的比酶活相近,都约为1.01×104U/mg;当添加不同浓度的丝氨酸后,野生酶的IC50值为7 μmol/L,即野生酶在丝氨酸浓度达到7 μmol/L时,酶活就被抑制为原来的50%,而突变酶 (N346A/H344A) 在丝氨酸浓度达到160 mmol/L时酶活仍保持96%。与野生型相比,本实验所构建突变体在解除了终产物L-丝氨酸对PGDH酶反馈抑制的同时,基本保持了原有野生型酶活性。
2.4野生型及突变型PGDH理化性质比较
酶液在待测温度下先预热1 min后加入同样温度下预热2 min的反应液中,测定不同反应温度下酶液的酶活。由曲线数据显示,野生型PDGH的最适温度为50 ℃,突变体的最适温度为55 ℃,高于野生型PDGH (图5A)。
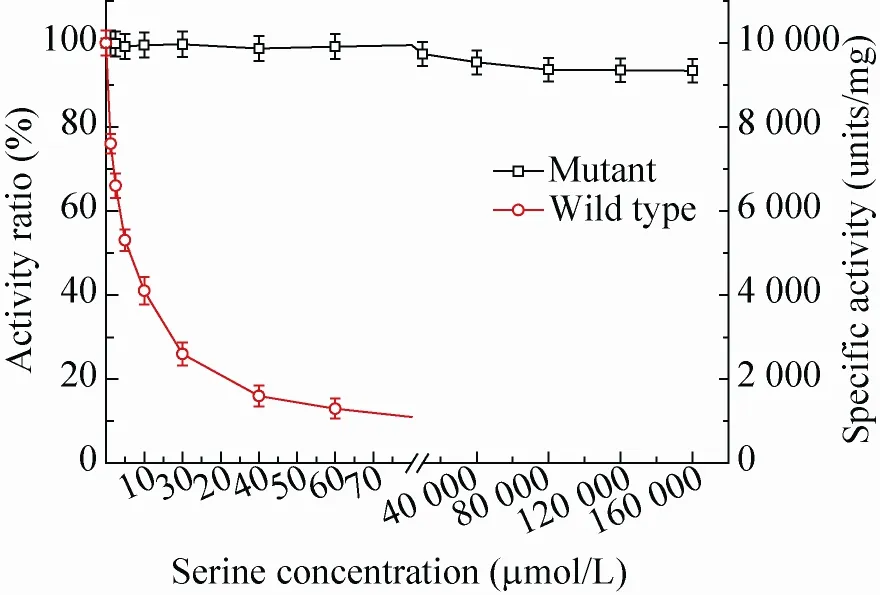
图4 野生型PGDH酶及其突变体 (N346A/H344A)的抗反馈抑制曲线Fig. 4 Feedback inhibition of wild-type PGDH and its mutant (N346A/H344A).

图5 野生型PGDH及突变体 (N346A/H344A) 理化性质比较Fig. 5 Compare of physicochemical property of wld type PGDH and its mutant (N346A/H344A). (A) Optimum temperature. (B) Thermostability. (C) Optimal pH. (D) pH-stability.
酶液在设定温度下保存3 min后置于冰上,测定在37 ℃反应温度下酶液的酶活。由曲线数据显示 (图5B),突变体和野生型PDGH的在0−30 ℃保存3 min后,酶活一直很稳定,但野生型PDGH在60 ℃保存3 min后迅速失活到原活性的7%,而突变体在60 ℃保存3 min后,仍保持原酶活的38%。由此得出突变体的最适温度和热稳定性都略高于野生型PDGH。
酶液被调节到不同的待测pH值后加入同pH值的反应液中,测定不同pH值下酶液的酶活。曲线数据显示 (图5C),突变体和野生型PDGH的最适pH值相当,都为pH 8.5,但在磷酸钾-钠缓冲液中酶活较同样pH值下 (pH 7.5)的磷酸钾缓冲液要小约1 000 U,即在后者中更适合酶的活性表达;在酸碱稳定性测定中,可观测到突变体 (N346A/H344A) 和野生型PDGH的酸碱稳定性基本一致 (图5D)。
实验同时也进行了该调控结合域的3个结合位点的单点突变,即单突变H344A、N346A 和N364’A和其他两个双点突变,即双突变H344A和N364’A,N346A和N364’A并采用相同方法构建了相应的突变体,结果单点突变体和双点突变H344A-N364’A、N346A-N364’A的突变体的酶活与野生型相比无明显差异,而对单突变体对丝氨酸的IC50最大值为52 μmol/L,尚未解除丝氨酸反馈抑制,而双点突变H344A-N364’A、N346A-N364’A的突变体对丝氨酸的反馈抑制效果与双点突变H344AN346A的突变体效果相当,且催化活力基本一致,具体数据见表4。

表4 丝氨酸对野生型PGDH及其突变体的抑制效果Table 4 Inhibition of L-Serine on wild-type PGDH and its mutants
3 小结
国内外报道中,去除SerA的反馈抑制的方法,多采用尝试性的删除部分结构域,以降低反馈抑制效应,如Wendisch报道研究发现在谷氨酸棒状杆菌中3-磷酸甘油酸脱氢酶C端197个氨基酸缺失后能有效地解除反馈抑制,但比酶活从2.1 U/mg下降到1.3 U/mg,研究者同时发现该酶野生型为同源四聚体,而突变体为同源二倍体[16]。张绪梅通过对大肠杆菌SerA碱基序列和蛋白质结构分析,用PCR突变法构建C端缺失不同序列的突变酶,获得了具有抗反馈抑制性质的大肠杆菌磷酸甘油酸脱氢酶,酶活性测定结果表明,M1、M2蛋白酶均保持了原有的野生型磷酸甘油酸脱氢酶活性,且部分解除了终产物L–丝氨酸的反馈抑制作用;M3蛋白酶完全解除了终产物的反馈抑制作用,但酶活为野生型的83%,且在实验过程中研究者发现去除部分结构域的菌株比野生型更易形成包涵体[17]。蒋琼等构建了甲基营养菌serA基因的缺失C末端ACT功能域的突变基因和重组体表达菌MB200/pCM80serA△ 77。分析发现野生型基因重组表达菌的PGDH酶活受L-丝氨酸浓度影响较大,当反应体系中的L-丝氨酸浓度为40 μmol/L时,酶活减少了约1/3;而缺失了ACT功能域的基因表达菌的PGDH酶活基本不变,不受L-丝氨酸浓度的影响[18]。除蒋琼的结果较为理想外,其他研究者通过非理性尝试删除部分调控域以达到解除反馈抑制并能保持酶活力不降的效果均不是很理想。值得注意的是,Al-Rabiee等也曾对来源于大肠杆菌Escherichia coli 的PGDH酶的相同位点进行过定点突变和动力学研究[6,19-21],其采用的表达载体为pTrc 99A,表达宿主菌为JM105,结果显示单突变体的丝氨酸IC50值与本实验接近,而双突变体的丝氨酸IC50的值均大于250 mmol/L,但实际活细胞中并不会在胞内出现浓度高达250 mmol/L的丝氨酸浓度,其突变体在丝氨酸浓度较低情况下的酶活保留率是未知的。相比而言,本实验明确给出双突变体 (N346A/H344A) 在丝氨酸浓度为160 mmol/L之前的抑制曲线图及突变体的酶学性质,同时本实验也明确给出不同突变体的动力学参数值,这对丝氨酸及其衍生物的代谢调控和生产实践将有更多的参考价值。
本研究在分析大肠杆菌SerA蛋白晶体结构的基础上,通过理性设计突变替换丝氨酸分子与酶分子调控域上直接结合的氨基酸残基分子为丙氨酸,阻断丝氨酸分子与酶分子的极性共价结合,希望获得解除丝氨酸对PDGH反馈抑制作用且酶活基本保持不变的突变体,实验研究结果表明上述目的达到,并且通过进一步的酶学特性和动力学实验表明,突变体和野生型在理化性质和催化活性上均无显著差别,而且实验中野生型和突变体的过量表达均没有产生明显的包涵体或聚集体,即选择的表达载体和表达宿主适合过量表达目的蛋白。此研究提高了L-丝氨酸中关键酶基因的表达量,并解除了产物的反馈抑制作用,为应用代谢工程方法研究L-丝氨酸的合成及其衍生物的合成提供了重要参考。
REFERENCES
[1] Snell K, Natsumeda Y, Weber G. The modulation of serine metabolism in hepatoma 3924A during different phases of cellular proliferation in culture. Biochem J, 1987, 245(2): 609–612.
[2] Grant GA, Hu Z, Xu XL. Specific interactions at the regulatory domain-substrate binding domain interface influence the cooperativity of inhibition and effector binding in Escherichia coli D-3-phosphoglycerate dehydrogenase. J Biol Chem, 2001, 276(2): 1078–1083.
[3] Schuller DJ, Grant GA, Banaszak LJ. The allosteric ligand site in the Vmax-type cooperative enzyme phosphoglycerate dehydrogenase. Nat Struct Biol, 1995, 2(1): 69–76.
[4] Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Vol. 3. New York: Cold Spring Harbor Laboratory Press, 1989: 908.
[5] Liu S, Hu BC. Strategy of protein soluble expression in Escherichia coli. Lett Biotech, 2005, 16(2): 172–175 (in Chinese).刘爽, 胡宝成. 原核系统可溶性表达策略. 生物技术通讯, 2005, 16(2): 172–175.
[6] Zhao G, Winkler ME. A novel alpha-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Escherichia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria. J Bacteriol, 1996, 178(1): 232–239.
[7] Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry, 1976, 72(1/2): 248-254.
[8] Tabor S, Richardson CC. A bacteriophage T7 RNApolymerase/promoter system for controlled exclusive expression of specific genes. Proc Natl Acad Sci USA, 1985, 82(4): 1074–1078.
[9] McKitrick JC, Pizer LI. Regulation of phosphoglycerate dehydrogenase levels and effect on serine synthesis in Escherichia coli K-12. J Bacteriol, 1980, 141(1): 235–245.
[10] Blomberg A, Adler L. Roles of glycerol and glycerol-3-phosphate dehydrogenase (NAD+) in acquired osmotolerance of Saccharomyces cerevisiae. J Bacteriol, 1989, 171(2): 1087–1092.
[11] Chen XZ, Fang HY, Rao ZM, et al. Cloning and characterization of a NAD+-dependent glycerol-3-phosphate dehydrogenase gene from Candida glycerinogenes, an industrial glycerol producer. FEMS Yeast Res, 2008, 8(5): 725–734.
[12] Albertyn J, Van Tonder A, Prior BA. Prior purification and characterization of glycerol-3-phosphate dehydrogenase of Saccharomyces cerevisiae. FEBS Lett, 1992, 308(2): 130–132.
[13] Grant GA, Xu XL, Hu ZQ. Quantitative relationships of site to site interaction in Escherichia coli D-3-phosphoglycerate dehydrogenase revealed by asymmetric hybrid tetramers. J Biol Chem, 2004, 279(14): 13452–13460.
[14] Grant GA, Hu ZQ, Xu XL. Cofactor binding to Escherichia coli D-3-phosphoglycerate dehydrogenase induces multiple conformations which alter effector binding. J Biol Chem, 2002, 277(42): 39548–39553.
[15] Thompson JR, Bell JK, Bratt J, et al. Vmaxregulation through domain and subunit changes. The active form of phosphoglycerate dehydrogenase. Biochemistry, 2005, 44(15): 5763–5773.
[16] Peters-Wendisch P, Netzer R, Eggeling L, et al. 3-Phosphoglycerate dehydrogenase from Corynebacterium glutamicum: the C-terminal domain is not essential for activity but is required for inhibition by L-serine. Appl Microbiol Biotechnol, 2002, 60(4): 437–441.
[17] Zhang XM, Guo CJ, Liu Y, et al. Construction and characterization of E. coli PGDH mutants with feedback-inhibition resistance. Chin J Biochem Mol Biol, 2006, 22(10): 806–810 (in Chinese).张绪梅, 郭长江, 刘云, 等. 大肠杆菌PGDH末端缺失突变体的构建及抗反馈抑制效应分析. 中国生物化学与分子生物学报, 2006, 22(10): 806–810.
[18] Jiang Q, Ma S, Zhou K, et al. Function of serA gene in M. sp. MB200. Genomic Appl Biol, 2014, 33(3): 551–555 (in Chinese).蒋琼, 马晟, 周侃, 等. 甲基营养菌M. sp. MB200中serA基因功能初探. 基因组学与应用生物学, 2014, 33(3): 551–555.
[19] Al-Rabiee R, Lee EJ, Grant GA. The mechanism of velocity modulated allosteric regulation in D-3-phosphoglycerate dehydrogenase cross-linking adjacent regulatory domains with engineered disulfides mimics effector binding. J Biol Chem, 1996, 271(22): 13013–13017.
[20] Grant GA, Schuller DJ, Banaszak LJ. A model for the regulation of D-3-phosphoglycerate dehydrogenase, a Vmax-type allosteric enzyme. Protein Sci, 1996, 5(1): 34–41.
[21] Schuller DJ, Fetter CH, Banaszak LJ, et al. Enhanced expression of the Escherichia coli serA gene in a plasmid vector. Purification, crystallization, and preliminary X-ray data of D-3 phosphoglycerate dehydrogenase. J Biol Chem, 1989, 264(5): 2645–2648.
(本文责编陈宏宇)
工业生物技术
Construction and characterization of Escherichia coli D-3-phosphoglycerate dehydrogenase mutants with feedback-inhibition relief
Hui Deng1,2, Cunwu Chen1, Chuanbo Sun1, and Chuanbao Wei1,2
1 College of Biology and Pharmaceutical Engineering, West Anhui University, Liu’an 237012, Anhui, China 2 Research Center of Protein Separation and Purification, Liu'an 237012, Anhui, China
Abstract:3-Phosphoglycerate dehydrogenase (PGDH, EC 1.1.1.95) is the key enzyme in L-serine biosynthesis and its coding gene is serA. PGDH is feedback inhibited by L-serine. In order to relieve the feedback-inhibition of PGDH by L-serine, H344 or D346 or D364 were chosen for site directed mutagenesis. The mutants were generated by the standard QuikChange mutagenesis, further subcloned into expression vector pT7-7 and transformed into Escherichia coli BL21 (DE3) cells. The recombinant cells were collected after cultured in LB media post induced by isopropyl beta-D-thiogalactopyranoside. The enzymes were purified by anion exchange chromatography, and SDS-PAGE showed that the purified enzymes were homogenous. Enzyme characterization indicated that the mutant enzyme showed similar activity, optimal temperature, and optimal pH as that of the wild-type enzyme. Moreover, feedback inhibition study showed that the activity of the double mutant (N346A/H344A) could remain 96% in the presence of serine up to 160 mmol/L, whereas the activity of the wild-type enzyme remains only 50% in the presents of serine of 7 μmol/L, thus successfully relieving the feedback inhibition of PGDH with its activity remained.
Keywords:Escherichia coli phosphoglycerate dehydrogenase, construction of mutants, relief of feedback inhibition
DOI:10.13345/j.cjb.150343
Corresponding authors: Chuanbao Wei. Tel/Fax: +86-564-3305073
