ABAB型酞菁衍生物取代基对结构和电子光谱的影响
2016-06-30王加亮阚玉和
王加亮, 李 晴, 阚玉和
(淮阴师范学院 化学化工学院, 江苏 淮安 223300)
ABAB型酞菁衍生物取代基对结构和电子光谱的影响
王加亮, 李晴, 阚玉和
(淮阴师范学院 化学化工学院, 江苏 淮安223300)
摘要:采用DFT方法在B3LYP/6-31G*水平上比较了ABAB型酞菁衍生物的几何结构,在此基础上用TDDFT方法计算了激发态,研究了ABAB型酞菁具有不同取代基对其电子结构和电子吸收光谱的影响. 结果表明,3,6-(3′,5′-双三氟甲基/三甲基)苯基的加入,前线轨道能级裂分导致Q带红移并出现了不同程度的裂分,Qx吸收带均红移,三氟甲基使Qy带蓝移. 这种大的取代基在可见光区谱带的调制功能可以有效提高敏化剂的光捕获效率,为新型高效染料分子的设计提供理论指导.
关键词:染料敏化剂; 酞菁; 含时密度泛函; 电荷差分密度; 电子吸收光谱
0引言
染料敏化太阳能电池(DSSCs)因其生产成本低、制造工艺相对简单以及可调的光学特性而获得了广泛的关注[1-4].目前研究的光敏染料种类众多,诸如多吡啶金属系列染料[5]、纯有机染料[6]以及卟啉和酞菁系列染料[7].其中,多吡啶钌配合物是最早应用于DSSCs且应用较多的的一类光敏染料,其光电转换效率在全光照条件下目前已突破11.3%[8],但由于多吡啶钌类染料在近红外区缺乏强的吸收带,从而限制了其光电转换效率的进一步提高.
近年来,新型的具有给体-共轭桥-受体(D-π-A)结构的有机染料异军突起[9],这类染料的特点是最高占据轨道(HOMO)集中在给体部分,最低未占轨道(LUMO)集中在受体部分给体与受体之间由共轭桥连接,由于受体与TiO2直接相连,使得激发电子能顺利地自受体注入TiO2的导带.酞菁类染料因其在紫外-可见光区具有强吸收带以及较高摩尔消光系数,吸引了世界上众多研究者的关注.目前,卟啉类共敏化DSSCs光电转换效率已突破13%[10],而酞菁作为染料敏化剂的光电转换效率最高仅有6.1%[11],造成这种低效率的主要原因是酞菁类染料分子在TiO2表面有强烈聚集作用[12].因此,合成具有非对称且有较大空间位阻取代基的酞菁衍生物成为目前的热点.最近,Torres等合成了碘代的ABAB型大空间取代基的3,6-(3′,5′-双三氟甲基)苯基酞菁[13],实验测得其在675和690 nm显示两个劈裂的Q带吸收峰.本文以此体系为模型,考虑取代基电子效应,设计了研究体系的几何结构(图1),分析不同偶极方向电子结构和电子光谱特征,为设计高效的D-π-A型染料提供重要的参考依据.
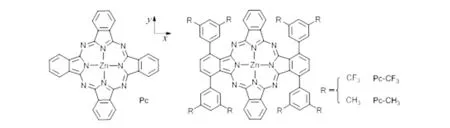
图1 研究体系的几何结构
1计算方法
所有计算采用Gaussian 09W程序包,几何优化在限制对称性条件下,采用杂化密度泛函B3LYP[14]在6-31G*基组水平下完成,所有优化的几何结构经频率计算验证无虚频均为稳定结构.基于含时密度泛函理论(TD-DFT)方法[15],为和实验对比,在长程校正密度泛函CAM-B3LYP[16]在6-31G*水平下完成电子吸收光谱的计算,溶剂化效应采用连续极化模型IEF-PCM[17]方法,溶剂为四氢呋喃(THF).电荷差分密度方法用来分析激发态电子跃迁特征.对于各种异构体,其结构权重的玻尔兹曼分布采用SpecDis程序[18]获得.
2结果讨论
2.1几何结构
考虑分子对称性形成的同分异构体,以三氟甲基取代基的ABAB型酞菁衍生物Pc-CF3为例,我们对7种具典型构象的异构体进行几何优化,其中C1和D2点群结构分别对应2种异构体,优化后的体系相关能量见表1.
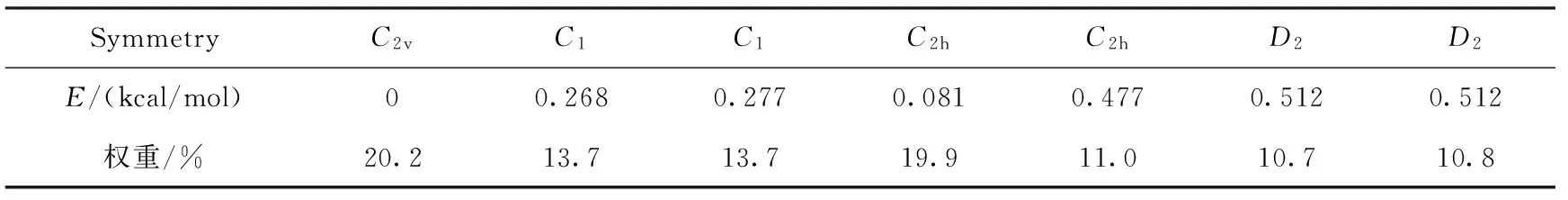
表1 三氟甲基酞菁衍生物的几种立体异构体的相对能量
从表1中几种结构的相对能量可以看出,C2v构型的能量最低,最不稳定的是D2,两者能量差为0.512 kcal/mol,能量权重的波尔兹曼分析表明,C2v构型和C2h-para构型所占的权重较大,分别为20.2%和19.9%.因此,下文讨论均取能量最低的C2v结构进行讨论.
2.2前线轨道
前线分子轨道作为电子结构性质研究的重要部分,对探讨激发态性质和电子吸收光谱等有重要作用.采用B3LYP/6-31G*方法计算了锌酞菁Pc及2种衍生物的前线分子轨道能级,图2对比列出了3种化合物的分子轨道能级.从图2中可以看出,具有D2h对称性的锌酞菁,其LUMO和LUMO+1是简并的,随着对称性的降低,Pc-CF3中这两个能级却出现了明显的裂分,相比较而言,甲基衍生物Pc-CH3的LUMO和LUMO+1虽然裂分,但并没有Pc-CF3明显,仍接近简并.另外,Pc-CF3的HOMO和LUMO能级较酞菁母体的HOMO和LUMO能级都有明显的降低,而LUMO能级比HOMO能级降低的更多,导致Pc-CF3的HOMO和LUMO间能隙有所降低.而Pc-CH3的HOMO和LUMO能级较锌酞菁母体Pc的HOMO、LUMO能级都有所升高,HOMO比LUMO增加的更多,导致Pc-CH3的HOMO、LUMO能隙较酞菁也有所降低.总的来说,吸电子基CF3的加入使酞菁原有的HOMO和LUMO轨道能量有所降低,给电子基的影响刚好相反.
2.3吸收光谱
考虑溶剂化效应采用含时密度泛函TD-CAM-B3LYP/6-31G*方法,计算激发态的电子跃迁,表2~4分别列出了Pc、Pc-CF3以及Pc-CH3三种化合物C2v构型(其它构象的光谱基本重合)的主要激发态电子跃迁、振子强度、跃迁偶极矩以及各激发态主要轨道贡献,并对其特征吸收Q带和B带涉及的主要跃迁本质做了进一步指认.
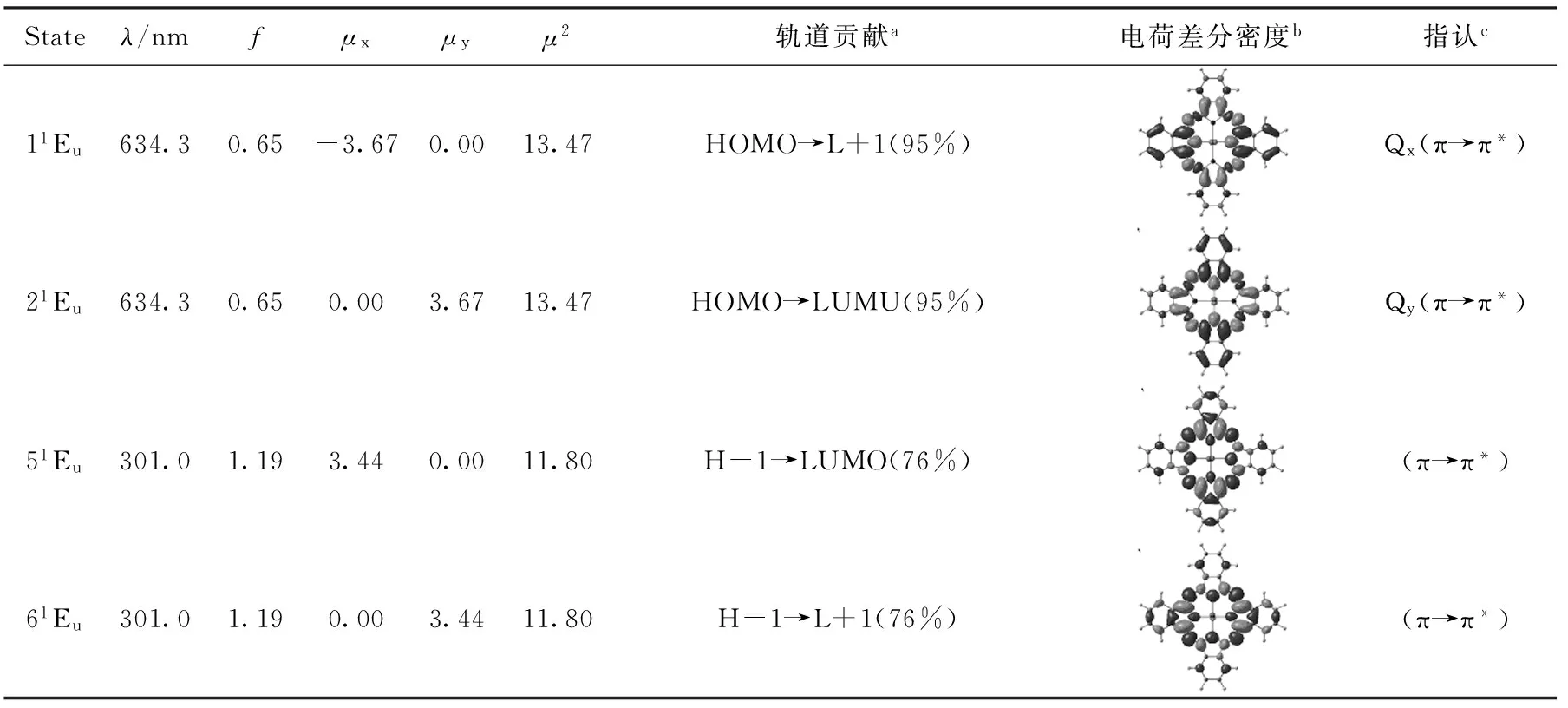
表2 Pc的主要激发态的跃迁能和振子强度(f)、跃迁偶极矩(μ)及跃迁指认
aR表示取代基;b黑色表示空穴,灰色表示电子;cH,L分别表示HOMO和LUMO.

表3 三氟甲基衍生物Pc-CF3的主要激发态的跃迁能和振子强度(f)、跃迁偶极矩(μ)及跃迁指认
aR表示取代基;b黑色表示空穴,灰色表示电子;cH,L分别表示HOMO和LUMO.
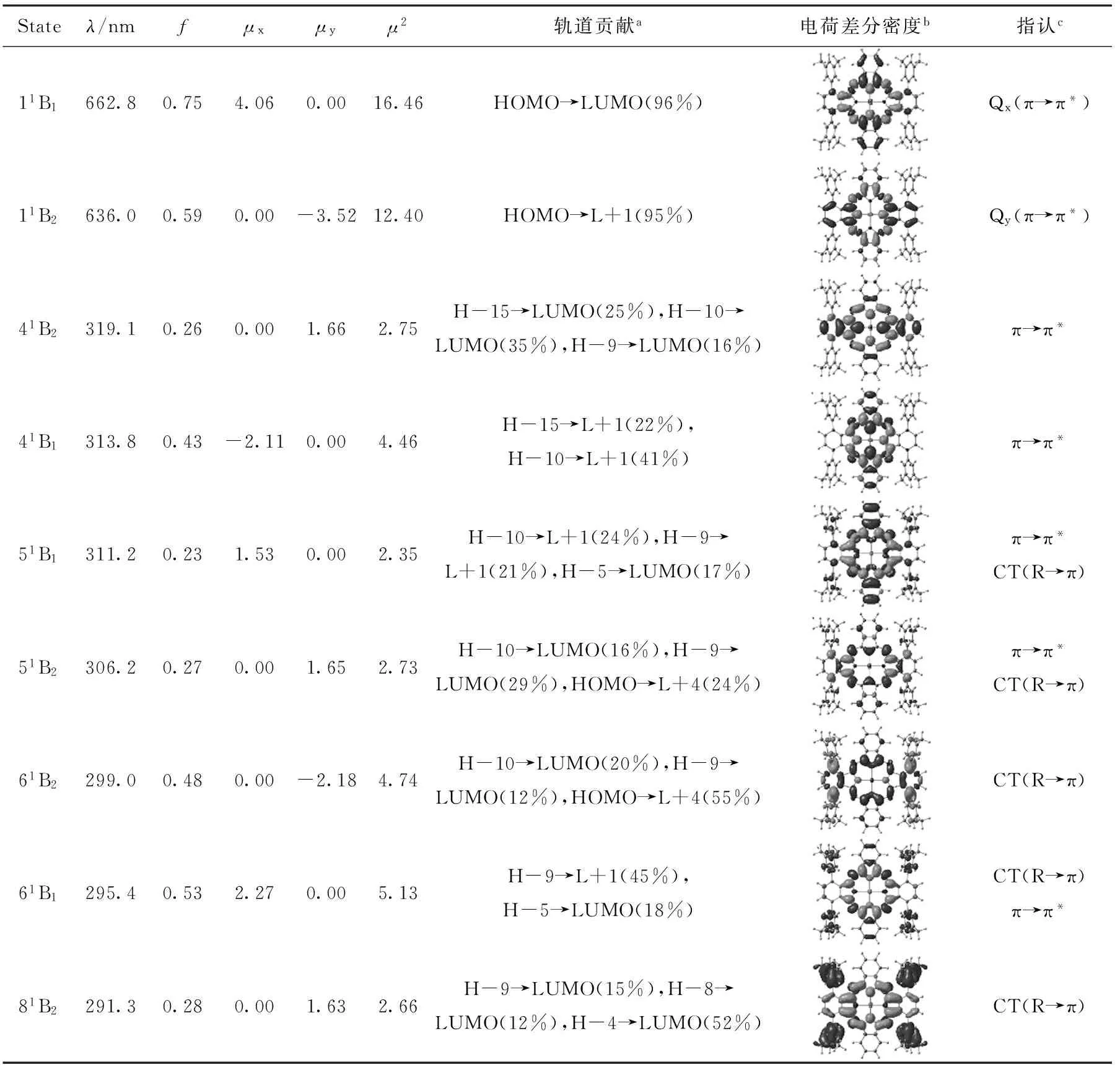
表4 甲基衍生物Pc-CH3的主要激发态的跃迁能和振子强度(f)、跃迁偶极矩(μ)及跃迁指认
aR表示取代基;b黑色表示空穴,灰色表示电子;cH,L分别表示HOMO和LUMO.
通常,酞菁类化合物在可见光区(500~750 nm)和紫外可见光区(200~400 nm)的强吸收带分别称为Q带、B带,而Q带因跃迁偶极矩分量(如图标识坐标方向)贡献不同,可分为Qx和Qy.计算拟合的
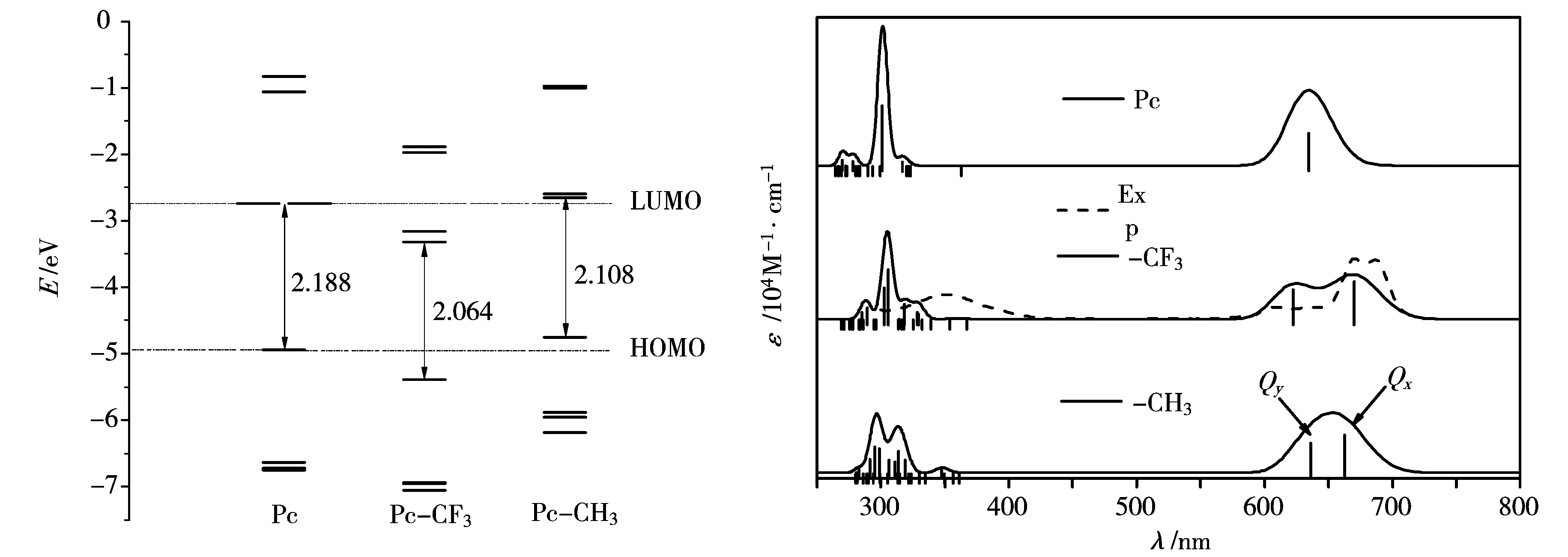

图2 Pc与两种ABAB型酞菁 衍生物的前线轨道能级比较图3 Pc及ABAB型酞菁衍生物在 THF溶剂中的紫外-可见吸收光谱
锌酞菁Pc、Pc-CF3和Pc-CH3三种化合物的电子吸收光谱列于图3,可以看出,计算结果与文献中实验值拟合较好.下面分别对Pc、Pc-CF3和Pc-CH3三种化合物的两个特征吸收谱带进行指认.
2.3.1Q带
计算的锌酞菁母体的Q带由两个简并的吸收峰(λ=634.3 nm)组成,与文献值[19]相比较为接近,分别对应11Eu和21Eu两个激发态,分别由HOMO→L+1(95%)、HOMO→LUMO(95%)的跃迁贡献,因此Q带激发态跃迁仅涉及Gouterman轨道.随着3,6-(3′,5′-双三氟甲基)苯基的加入,可以观察到锌酞菁母体原本简并的Qx和Qy带出现了明显裂分,激发态的对称性也明显降低,分别在670.1和622.6 nm处出现了两个吸收峰,即Qx处吸收峰相对于锌酞菁母体在该处的吸收发生了较明显的红移,而Qy处略微蓝移.这可以从前线轨道能级得到解释,因为Pc-CF3的Qx和Qy分别来自HOMO→LUMO和HOMO→L+1的贡献(表3),而HOMO和LUMO能隙较Pc母体降低,并且LUMO和L+1能级发生了明显的裂分,前者直接导致了Qx峰的红移,后者导致了Qy峰的蓝移,从而使Qx和Qy裂分.而对于Pc-CH3的Q带,也观察到同样裂分的现象,与Pc-CF3不同的是,Pc-CH3的Qx和Qy带较Pc而言都呈红移,Qy带红移主要是由于HOMO能级相对增加而LUMO和LUMO+1近似简并,即HOMO→L+1跃迁能较小导致的.基态和激发态电荷差分密度分析表明,取代基未改变Q带的π→π*跃迁本质.此外,对比表2~4,可以观察到Qx跃迁的振子强度较酞菁母体有所提高,而Qy跃迁的振子强度略有降低,这与x、y方向跃迁偶极矩μ大小相对应.因此,对本文讨论的ABAB型酞菁衍生物,R取代基的加入,有利于增强和宽化Q吸收带,这有利于提高该类化合物作为有机染料敏化剂的在可见光区的光捕获效率.
2.3.2B带
对于Pc来说,B吸收带比较典型的是在301.0和269.8 nm处的两个峰,分别对应两对简并的激发态51Eu、61Eu和91Eu、101Eu.电荷差分密度分析表明,主要为π→π*跃迁贡献.而对两种取代衍生物,由于对称性的降低,B带宽化,除了前线轨道跃迁贡献外,取代基贡献轨道致使电子跃迁更加复杂.
图3中可以看出,Pc-CF3主要由三个吸收带组成,一处强的、两处相对较弱的,与Pc的B带峰形较为接近.分析表3中的计算结果,B带最强的吸收峰主要来自于305.9(51B1)、302.8(61B2) nm这两处跃迁的贡献,对应的振子强度分别为0.99和0.63.电荷差分密度分析表明,51B1态跃迁的贡献主要来自H-10→LUMO,H-3→L+1跃迁,而302.8 nm处的61B2态跃迁主要来自H-7→LUMO和H-5→LUMO跃迁贡献,电荷差分密度图表明二者皆以π→π*为主.最强吸收峰左侧的吸收峰主要来自61B1和71B2跃迁,这两个态跃迁虽然包含组态较为复杂,但通过电荷差分密度图可以看到其跃迁主要为π→π*跃迁.中间最强吸收带右侧吸收带主要由31B1、31B2、41B2三个态的跃迁贡献,分析计算结果发现,31B1的跃迁除了H-10→LUMO的π→π*的定域激发跃迁贡献外,存在明显的H-2→LUMO(57%)贡献的取代基到酞菁环(R→π的电荷转移跃迁,而31B2态的跃迁则以酞菁环到取代基(π→R)的电荷转移跃迁为主.41B2的跃迁所涉及的跃迁组态更为复杂,但主要为π→π*跃迁,还有少量π→R和R→π的电荷转移跃迁.
与Pc和PcCF3不同,PcCH3的B带在300 nm左右呈现两个强度相当的吸收峰,对于大于300 nm吸收峰,分析其振子强度较大的跃迁态41B1、41B2、51B1和51B2,主要为π→π*定域激发跃迁,而在小于300 nm处吸收峰,贡献相对较大的61B2、61B1和81B2态的电荷差分密度分析可以明显指认,该处吸收主要为R→π和π→R的电荷转移跃迁贡献.
3结论
采用密度泛函理论方法在B3LYP/6-31G*水平上优化ABAB型锌酞菁衍生物几何结构,并用TDDFT方法在同样水平下计算了激发态,研究了不同取代基对其电子结构以及电子吸收光谱的影响. 结果表明,取代基的加入,破坏了分子的对称性,改变了前线分子轨道能级,导致酞菁Q带出现了明显的裂分,Qx发生红移;对于Qy带,三氟甲基衍生物呈现蓝移,而甲基衍生物略微红移.取代基使酞菁衍生物的B带宽化,并存在明显的电荷转移跃迁.这种大的取代基在可见光区谱带的调制功能可以有效提高敏化剂的光捕获效率,为理性设计染料敏化太阳能电池敏化剂及实验合成提供有益的参考.
参考文献:
[1]O'Regan B, Gratzel M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films[J]. Nature, 1991, 353: 737-740.
[2]王红军, 王艳蕊, 关梦杰, 等. 染料敏化纳米晶薄膜太阳能电池用染料敏化剂的研究进展[J]. 化学工程师, 2010(10): 22-26.
[3]杨振清, 曹达鹏, 邵长金, 等. 染料敏化太阳电池中有机染料敏化剂的研究进展[J]. 中国材料进展, 2011(12):46-51.
[4]何俊杰, 陈舒欣, 王婷婷, 等. 有机染料敏化剂分子设计新进展[J]. 有机化学, 2012, 32: 472-485.
[5]周迪, 佘希林, 宋国君. 金属有机类光敏剂在染料敏化太阳能电池中的应用[J]. 贵金属, 2010, 31(1):37-42.
[6]武国华, 孔凡太, 翁坚,等. 有机染料及其在染料敏化太阳电池中的应用[J]. 化学进展, 2011(9):1929-1935.
[7]李孜, 贾春阳, 万中全. 卟啉类光敏剂在染料敏化太阳能电池中的应用[J]. 化学进展, 2011, 23(5):1014-1021.
[8]Gao F, Wang Y, Shi D, et al. Enhance the Optical Absorptivity of Nanocrystalline TiO2 Film with High Molar Extinction Coefficient Ruthenium Sensitizers for High Performance Dye-Sensitized Solar Cells[J]. J Amer Chem Soc, 2008, 130(32):10720-10728.
[9]谢小银, 栾国颜. D-π-A型染料敏化太阳能电池有机敏化分子设计研究进展[J]. 吉林化工学院学报, 2015(11):12-15.
[10]Mathew S, Yella A, Gao P, et al. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers[J]. Nat Chem, 2014, 6(3): 242-247.
[11]Ragoussi M E, Cid J J, Yum J H, et al. Carboxyethynyl anchoring ligands: a means to improving the efficiency of phthalocyanine-sensitized solar cells[J]. Angew Chem Int Ed, 2012, 51(18): 4375-4378.
[12]Singh V K, Kanaparthi R K, Giribabu L. Emerging molecular design strategies of unsymmetrical phthalocyanines for dye-sensitized solar cell applications[J]. Rsc Adv, 2013, 4(14):6970-6984.
[13]Fazio E, Jaramillo-Garcia J, Torres T. Efficient synthesis of ABAB functionalized phthalocyanines[J]. Org Lett, 2014, 16(18): 4706-4709.
[14]Becke A D. Density functional thermochemistry: The role of exact exchange[J]. J Chem Phys, 1993, 98(7): 5648-5652.
[15]Bauernschmitt R, Ahlrichs R. Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory[J]. Chem Phys Lett, 1996, 256(4/5): 454-464.
[16]Yanai T, Tew D P, Handy N C. A new hybrid exchange-correlation functional using the Coulomb-Attenuating Method(CAM-B3LYP)[J]. Chem Phys Lett, 2004, 393(1/3):51-57.
[17]Mennucci B, Cammi R, Tomasi J. Excited states and solvatochromic shifts within a nonequilibrium solvation approach: A new formulation of the integral equation formalism method at the self-consistent field, configuration interaction, and multiconfiguration self-consistent field level[J]. J Chem Phys, 1998, 109(7):2798-2807.
[18]Bruhn T, Schaumloffel A, Hemberger Y, et al. SpecDis: Quantifying the Comparison of Calculated and Experimental Electronic Circular Dichroism Spectra[J]. Chirality, 2013, 25(4): 243-249.
[19]Rio Y, Rodriguez-Morgade M S, Torres T. Modulating the Electronic Properties of Porphyrinoids: A Voyage from the Violet to the Infrared Regions of the Electromagnetic Spectrum[J]. Org & Biomol Chem, 2008, 39(36):1877-1894.
[责任编辑:蒋海龙]
The Effects of Different Substituents on the Structure and Electronic Spectrum of ABAB-type Phthalocyanine
WANG Jia-liang, LI Qing, KAN Yu-he
(School of Chemistry and Chemical Engineering, Huaiyin Normal University, Huaian Jiangsu 223300, China)
Abstract:The molecular geometries, electronic structures of ABAB-type Zn(II) phthalocyanine compounds were investigated using the B3LYP method within a framework of density functional theory (DFT). The excitation energies of these molecules were computed by the time-dependent DFT (TD-DFT) methods. The calculated results of three compounds reflect the interpolation of bulky 3,6-(3′,5′-bis(trifluoromethyl/methyl)phenyl)phthalonitrile results in a red shift of the Q band and a different splitting degree. The specific performance can be described as the red shift of their Qx band and the blue shift of Qy band of phthalocyanine derivatives with trifluorine methyl groups. Large substituents with adjustment function in the visible region can effectively improve the light harvesting efficiency of sensitizer, which provides theoretical guidance for design of high efficient dyes.
Key words:dye sensitizer; phthalocyanine; time-dependent density functional theory; charge difference density; electronic absorption spectra
收稿日期:2016-02-26
基金项目:江苏省自然科学基金资助项目(BK2011408); 江苏省高校大学生实践创新训练计划项目(201510323005Z)
通讯作者:阚玉和(1971-),男,吉林敦化人,教授,博士,研究方向为应用量子化学. E-mail: kyh@hytc.edu.cn
中图分类号:O641
文献标识码:A
文章编号:1671-6876(2016)02-0126-06
