蛋白质空间结构相似度多参数算法模型的建立
2016-06-27张萍萍张建华尹咪咪
张萍萍, 张建华, 尹咪咪
(郑州大学 电气工程学院 河南 郑州 450001)
蛋白质空间结构相似度多参数算法模型的建立
张萍萍, 张建华, 尹咪咪
(郑州大学 电气工程学院 河南 郑州 450001)
通过收集165对蛋白质的结构文件,利用BLASTP比较它们的相似度.建立球极坐标系,分别将球体半径、方位角和仰角二等分和三等分,将蛋白质划分为8块和27块类似球壳碎片的区域.在此基础上,利用MATLAB计算12个参数相似度,用SPSS建立了二等分和三等分时总相似度和12个参数相似度的全回归模型、逐步回归模型和相关性回归模型.利用MATLAB建立BP神经网络模型,并与线性回归模型进行了对比.根据二等分时逐步回归模型的结果可以看出,原子个数相似度,C、N原子个数相似度,P、S的位置相似度以及密度相似度和总体相似度的相关性最显著.二等分时结果较三等分时好,逐步回归模型的结果最好.
蛋白质; 相似度; 回归分析; 逐步回归; BP神经网络
0 引言
蛋白质是组成生物体的物质基础,是生物功能的体现者,了解蛋白质的三维结构能够有效地揭示蛋白质的功能.蛋白质的结构和功能是统一的,蛋白质的生物功能在很大程度上依赖于其空间结构[1],结构相似的蛋白质功能也往往相似.文献[2]提出了一种基于骨架的蛋白质三维模型形状相似性分析方法.文献[3]在扇形划分模型的基础上提出了一种基于方差和均值统计描述的蛋白质结构相似性比较方法.文献[4] 使用混沌游走方法比较蛋白质的相似度.文献[5]对Ca骨架进行同密度划分和同心球划分,提取VPT特征分别判定相似性.文献[6]对比较蛋白建立球极坐标系,比较各区域Ca的相似性,利用半径和首尾距离来比较蛋白质的相似度.本文在上述研究基础上,计算各区域密度相似度,P、S个数和位置相似度,原子个数相似度,氨基酸个数和种类相似度,C、N、O、H原子个数相似度,然后运用统计学方法建立它们与总相似度之间的线性关系,找出与总相似度相关性最显著的特征.采用BP算法建立学习网络计算总体相似度,并与线性回归模型进行了对比.
1 方法
1.1 蛋白质的结构划分
在文献[7]基础上建立蛋白质的球极坐标系,不仅根据球体半径将蛋白质划分成等厚度球壳状,而且也等划分方位角和仰角,形成一个假想球壳碎片的空间划分方法.作为比较,分别将球体半径、方位角和仰角划分成二等分和三等分,也就是将蛋白质分为8块和27块来进行计算.
1.2 蛋白质数据的来源
选取一些常见的蛋白质,如S100家族、TNFa、RAS等,通过PDB数据库获取蛋白质编码,下载PDB文件,通过NCBI查找它们的氨基酸序列.将得到的氨基酸序列进行BLASTP找到与这些蛋白质相似的蛋白,下载这些相似蛋白的PDB文件,并标记每对蛋白的相似度,共找到了待分析的蛋白165对.
1.3 参数的选取
蛋白质结构复杂多变,不能通过单一参数来比较两个蛋白质的相似性,可以通过多参数分别建立线性与非线性关系来计算相似度.本文选取了12个参数,分别为原子个数相似度,氨基酸个数和种类相似度,C、N、O、H原子个数相似度,P、S个数和位置相似度以及密度相似度,所选取的参数都在一定程度上影响着蛋白质功能的发挥.参数的选取是经所有作者和几位生物学教授讨论决定的.
1.4 参数相似度的计算方法
1) 原子个数相似度(Sim1)
蛋白质内原子个数的多少决定了它的大小和质量,因此将蛋白质的原子个数相似度Sim1作为总相似度的一个参数.假设待比较蛋白为蛋白A和蛋白B,统计两个蛋白的原子总数分别为n1和n2,原子个数相似度计算方法如下:

(1)
2) 氨基酸个数和种类相似度(Sim2,Sim3)
氨基酸是蛋白质的基本组成单元,因此,统计蛋白A和蛋白B内氨基酸的个数和种类,计算它们的相似度,作为总相似度的一个参数.Sim2为氨基酸个数相似度,Sim3为氨基酸种类相似度.氨基酸个数和种类的相似度也可以采用式(1)计算.此时,n1为第1个蛋白的氨基酸个数和种类,n2为第2个蛋白的氨基酸个数和种类.
3) C、N、O、H原子个数相似度(Sim4~Sim7)
C几乎占了蛋白质成分的50%左右,文献[8]从蛋白质Ca骨架的角度出发,计算TM-score的值来比较结构的相似度.此外,N、O、H几乎占了蛋白质组成元素的40%以上,所以它们的相似性也作为总相似性的一个参数,比较方法依旧采用式(1).此时,n1为第一个蛋白的C、N、O、H的个数,n2为第二个蛋白的C、N、O、H的个数,Sim4为C原子个数相似度,Sim5为N原子个数相似度,Sim6为O原子个数相似度,Sim7为H原子个数相似度.
4) P、S个数和位置相似度(Sim8~Sim11)
S在蛋白质中的含量为0~3%,P的含量更少,但它们的存在会导致蛋白质的结构和功能发生大的变化,不同位置对蛋白质的结构和功能的影响也不一样,因此将P、S个数和位置相似度作为总相似度的一个参数.Sim8为P原子个数相似度,Sim9为P原子位置相似度,Sim10为S原子个数相似度,Sim11为S原子位置相似度.对P、S位置相似度的计算采用如下方法:因为P/S个数较少,首先根据球体半径等分后,看两个蛋白相同区域内是否含有P/S,若在同一块区域里面都含有或都不含有P/S,则这一块区域P/S的相似度为1,若一个区域含有P/S,另一个区域不含有P/S,则相似度为0.同理,计算将方位角和仰角等分后区域的 P/S相似度,这样可以得到每一块区域的P/S位置相似度,总的P/S位置相似度计算公式如下:
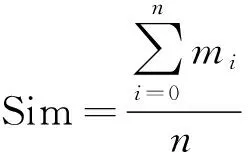
(2)
式中:mi为第i个分块区域的P/S相似度;n为分块个数.P/S个数相似度也可以采用式(1)计算.
5) 密度相似度(Sim12)
首先计算分块的每个区域的原子个数,比较第i个区域的原子个数相似度si,然后计算该区域在蛋白质中所占的比重li.si可以根据式(1)得到,li为

(3)
式中:n1i为第1个蛋白第i个区域的原子个数;n2i为第2个蛋白第i个区域的原子个数.则密度相似度计算公式如下:

(4)
式中:n为分块个数.
1.5 线性回归模型的建立
利用MTALAB可以得到每对相似蛋白的12个参数的相似度,通过BLASTP得到了每对蛋白的总体相似度,采用SPSS软件来分析这12个参数和总体相似度的关系.为了进行比较,分别在二等分和三等分时利用SPSS对Sim和Sim1~Sim12建立全回归模型,方法为“enter”;对Sim和Sim1~Sim12建立逐步回归模型,方法为“stepwise”;对Sim和Sim1~Sim12进行两两相关分析,并用和Sim相关性水平在 0.01内的参数建立相关性回归模型,方法为“enter”.
1.6 BP网络模型的建立
根据线性回归模型的建立比较两种划分区域方式,对结果比较好的一种建立BP神经网络模型,由于BP网络的学习和记忆具有不稳定性,故运行多次,找到比较好的一次结果,对网络进行保存,以便下次使用时调用.为了防止过学习,没有划分训练数据和测试数据,而是采用交叉验证的方式,提高了学习速率.
2 结果
2.1 线性回归模型
分别将球体半径、方位角和仰角划分成二等分和三等分区域,也就是将蛋白质分为8块和27块,利用SPSS软件建立165组对比蛋白的12个参数相似度和总相似度的线性回归模型,结果如表1所示.可以看出,这些模型的P都小于0.001,说明这些线性回归模型都极显著.二等分时逐步回归模型的R值最接近1,F值最大,误差最小,说明二等分时逐步回归模型的结果最好.此外,二等分时的结果要明显好于三等分时的结果.
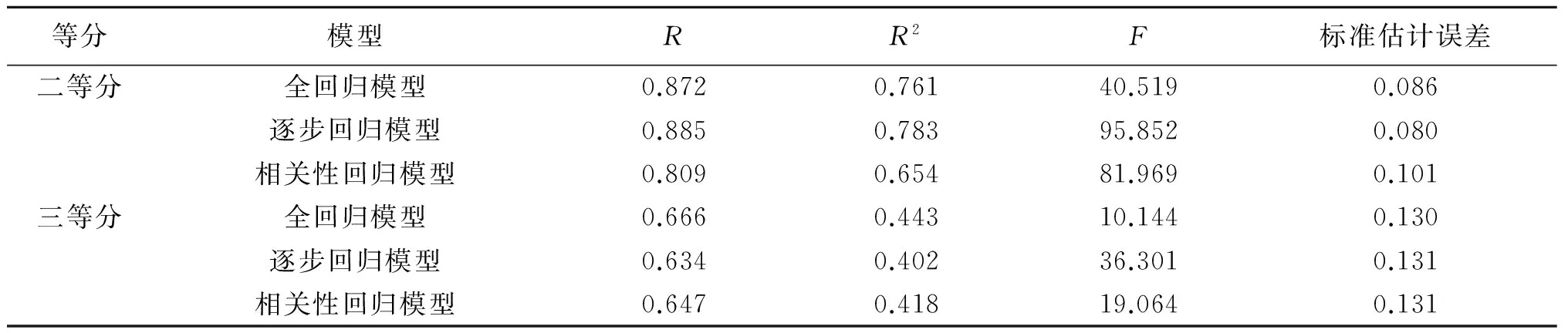
表1 线性回归模型结果
与BLASTP的相似度进行比较,线性回归模型的结果和误差如图1~4所示.二等分时实际误差:全回归模型为0.083 3,逐步回归模型为0.076 8,相关性回归模型为0.105 3.三等分时实际误差:全回归模型为0.139 8,逐步回归模型为0.143 7,相关性回归模型为0.142 0.可以看出,二等分时结果要比三等分时结果好,也就是将蛋白质分为8块区域时结果较好.
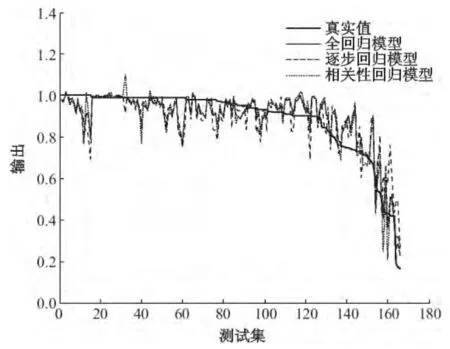
图1 二等分时线性回归模型的结果
Fig.1 Linear regression model results in bisection

图2 二等分时线性回归模型的误差
Fig.2 Linear regression model errors in bisection
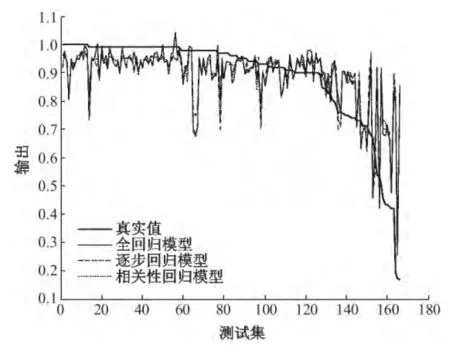
图3 三等分时线性回归模型的结果
Fig.3 Linear regression model results in trisection
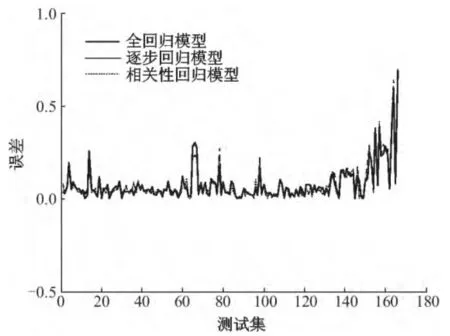
图4 三等分时线性回归模型的误差
Fig.4 Linear regression model errors in trisection
二等分时全回归模型公式为: Sim=0.691+1.027*Sim1-0.253*Sim2-0.143*Sim3-4.663*Sim4+2.521*Sim5+1.242*Sim6+ 0.012*Sim7-0.197*Sim8+0.44*Sim9+0.087*Sim10+0.101*Sim11+0.149*Sim12.
二等分时逐步回归模型公式为: Sim=0.362+0.936*Sim1-2.373*Sim4+1.566*Sim5+0.291*Sim9+0.110*Sim11+0.115*Sim12.
二等分时相关性回归模型公式为: Sim=-0.199+0.299*Sim1+0.143*Sim2+0.003*Sim8+0.388*Sim9+0.123*Sim10+0.073*Sim11+0.187*Sim12.
从二等分逐步回归模型的结果来看,原子个数相似度,C、N原子个数相似度,P、S位置相似度以及密度相似度和总体相似度的相关性最显著,用其建立的方程来比较蛋白质的相似度较另外两个模型效果更好.
2.2 BP神经网络模型
BP神经网络每次运行结果不一样,误差为0.023~0.34,选取误差为0.030 2时保存训练网络.BP神经网络模型的结果和误差如图5和图6所示,可以看出,BP神经网络的结果比线性回归模型的结果要好.
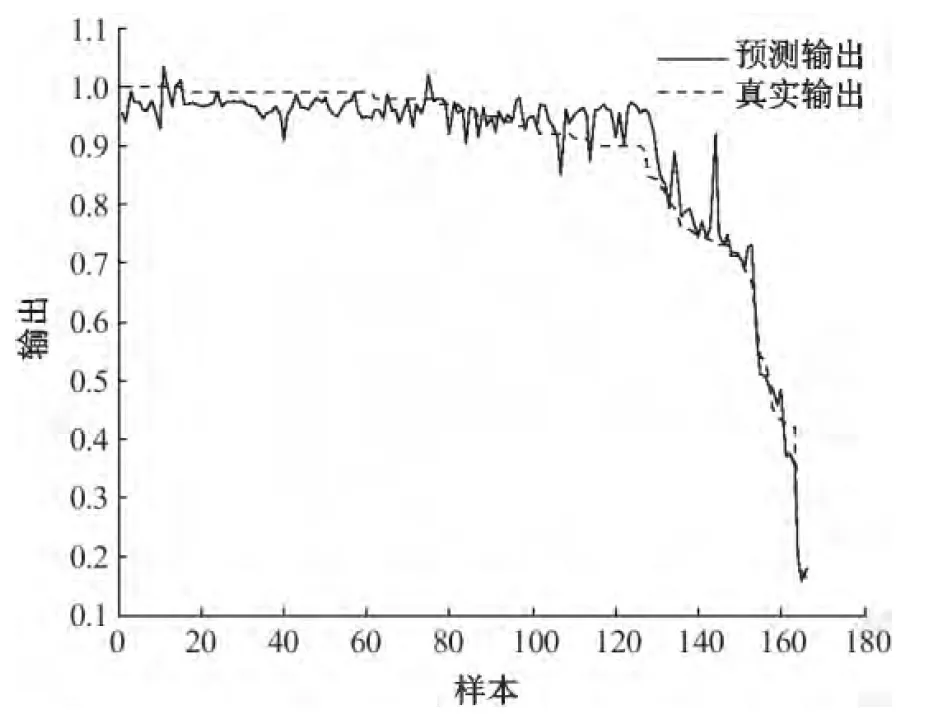
图5 BP神经网络模型的结果
Fig.5 Results of BP neural network model

图6 BP神经网络模型的误差
Fig.6 Errors of BP neural network model
3 讨论
介绍了比较蛋白质空间结构相似度模型的建立方法,首先找到待比较蛋白的坐标中心,建立球极坐标系,根据球体半径、方位角和仰角将蛋白质划分区域,形成一个假想球壳碎片的空间划分方法.作为比较,分别将球体半径、方位角和仰角划分成二等分和三等分,也就是将蛋白质分为8块和27块来进行计算.从线性回归模型的建立结果可以看出,二等分时的结果要明显好于三等分.3个线性回归模型中逐步回归模型的结果最好,误差最小.虽然这12个参数都对蛋白质的结构和功能有影响,但是在建立模型时,数据可能会有冗余,需要删去冗余项.根据二等分时逐步回归模型的结果可以看出,原子个数相似度,C、N原子个数相似度,P、S位置相似度以及密度相似度和总体相似度的相关性最显著.
作为比较,建立了BP神经网络模型来计算蛋白质的相似度,误差为0.023~0.34,保存了其中误差为0.030 2时的训练网络,可以作为以后比较蛋白的参考.这里只收集了165对蛋白的资料,今后还可以继续补充蛋白资料,以提高BP网络的准确性.虽然线性回归模型没有BP神经网络模型的效果好,但它是一个固定的数学式,可以很方便地得出两个蛋白的相似度,而BP神经网络结果不固定,每次训练还需要调用训练网络,因此两者各有所长.
[1] FATEMI M H,GHARAGHANI S.A novel QSAR model for prediction of apoptosis-inducing activity of 4-aryl-4-H-chromenes based on support vector machine[J].Bioorganic and medicinal chemistry,2007,15(24):7746-7754.
[2] LI Z,QIN S W,YU Z Y,et al.Skeleton-based shape analysis of protein models [J].Journal of molecular graphics and modelling,2014,53:72-81.
[3] 王雪平,王长缨.基于统计方法描述的蛋白质三维结构相似性比较[J].闽南师范大学学报(自然科学版),2014,27(1):39-43.
[4] 徐占.蛋白质空间结构的相似性比较[D].无锡:江南大学,2010.
[5] HOKSZA D.DDPIn-distance and density based protein indexing[C]// Proceeding of the 6th Annual IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology.Nashville, 2009:263-270.
[6] CHEN Z T.Analyzing influence on the conformation of single-chain antibody with the differential length of linkers[J].African journal of microbiology research,2011, 5(31):5737-5744.
[7] MARK J, IRENA Z, YAN R, et al. NCBI BLAST:a better web interface[J].Nucleic acids research,2008,36(12):5-9.
[8] HUNG L H,SAMUDRALA R.Accelerated protein structure comparison using TM-score-GPU[J].Bioinformatics,2012,28(16):2191-2192.
(责任编辑:孔 薇)
Establishing Algorithm Model of the Protein Spatial Structure Similarity Based on Multi-parameter
ZHANG Pingping, ZHANG Jianhua, YIN Mimi
(SchoolofElectricalEngineering,ZhengzhouUniversity,Zhengzhou450001,China)
One hundred and sixty-five pairs of protein structural files were collected and BLASTP was then utilized to compare their similarities. The spherical polar coordinate was established. The radius of the sphere, the azimuth and elevation were bisected and trisected, respectively, so the protein was divided into 8 and 27 blocks which were similar to spherical shell fragments. On this basis, the similarity of 12 parameters was calculated using MATLAB. The full regression model, stepwise regression model and filter regression model between the total similarity and the similarity of 12 parameters when they were bisected and trisected were established using SPSS. The BP neural network model was established using MATLAB for comparison. According to the results of stepwise regression model, similarity of the atomic number, similarity of C and N atomic number, similarity of P and S position and density had the most significant correlation with the overall similarity. Results of bisection were much better when compared with that of trisection,and stepwise regression model had the best results.
protein; similarity; regression analysis; stepwise regression; BP neural network
2015-10-07
国家自然科学青年基金资助项目(813D3150);中国中医药行业科研专项基金资助项目(201007001).
张萍萍(1991—),女,河南汝州人,硕士研究生,主要从事生物信息学研究,E-mail:385716230@qq.com;通讯作者:张建华(1971—),男,河北唐山人,副教授,博士研究生,主要从事生物医学信息的采集、分析及处理研究,E-mail:petermails@163.com.
张萍萍,张建华,尹咪咪. 蛋白质空间结构相似度多参数算法模型的建立[J]. 郑州大学学报(理学版),2016,48(2):105-109.
Q816
A
1671-6841(2016)02-0105-05
10.13705/j.issn.1671-6841.2015211
