炭/炭复合材料热解炭基体微观结构转变动力学
2016-06-20黄清波白瑞成李爱军孙晋良
黄清波, 张 丹, 白瑞成, 李爱军, 孙晋良
(1.上海大学 材料复合及先进分散技术教育部工程中心, 上海200072)(2.上海大学 上海市应用数学及力学研究所, 上海200072)
炭/炭复合材料热解炭基体微观结构转变动力学
黄清波1,张丹2,白瑞成1,李爱军1,孙晋良1
(1.上海大学 材料复合及先进分散技术教育部工程中心, 上海200072)(2.上海大学 上海市应用数学及力学研究所, 上海200072)
摘要:对热解炭沉积和织构形成过程进行动力学建模,重点分析C/C复合材料CVI制备工艺中基体炭形成时中织构/高织构(MT/HT)热解炭之间轮廓分明的急剧转变现象。基于Langmuir-Hinshelwood(L-H)理论和Particle-filler(P-F)概念模型,将MT和HT热解炭作为炭的两种亚稳相,以气相中占优的两种中间组分作为基体炭前驱体(线性小分子烃和小分子芳香烃),考虑基体表面单分子沉积形成MT热解炭和P-F双分子反应形成HT热解炭的过程,建立包含吸附/解吸附/脱氢的多步非均相热解炭沉积和织构形成反应动力学模型,研究该动力学系统达到稳态时热解炭随气相组成变化的情况。结果表明,热解炭沉积和织构形成过程曲线呈现“S”型特征,该曲线的线性稳定性分析表明热解炭沉积中的织构转变是一个包含迟滞区间的双稳态过程,进一步的计算表明此迟滞区间的大小明显受初始直链烃浓度以及沉积温度的影响。
关键词:炭/炭复合材料; 热解炭; 微观结构; 动力学
1前言
炭/炭(C/C)复合材料是一种高性能高温结构材料,具有高比强度/比模量、良好的韧性,在高温状态下具有优良的强度保持率,以及优良的摩擦性能和较高的导电、导热系数,广泛应用于航空、航天等领域。按照制备工艺不同,炭/炭复合材料的基体炭可分为:沥青炭、树脂炭和气相热解炭,以后者为基体的炭/炭复合材料具有更好的综合性能。等温等压化学气相渗透(I-CVI)工艺是制备热解炭基体最常用的工艺,其特征是采用气相烃类为前驱气体,经复杂的气相反应和表面沉积在炭纤维编制体内部得到热解炭基体。根据工艺条件不同,烃类气体的热解可以生成具有不同织构的热解炭基体,而不同的热解炭织构直接影响到材料宏观力学性能,尽管采用高分辨透射电子显微镜(HR-TEM)观察发现高织构热解炭(HT)内织构仍存在渐变的现象,但在偏光显微镜下观察经常发现不同织构间的变化是陡变的,其中最显著的是中织构(MT)和高织构(HT)热解炭之间的突变现象[1,2]。如图1所示,相比子图1(a,b)的单一织构占优情况,图1(c)中MT与HT热解炭分别主导沉积前期和后期,且相互之间泾渭分明,并没有显著的过渡区。因此,探索C/C复合材料中MT/HT热解炭突变动力学是深入理解热解炭的表面沉积和织构形成过程的钥匙,有助于对C/C复合材料热解炭微观结构的预测,并为最终实现热解炭微观结构的可控制备提供理论支持。
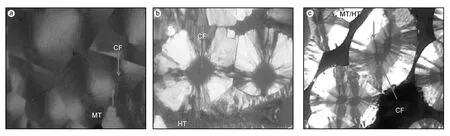
图 1 偏光显微镜下不同热解炭微观结构的光学活性:(a)MT,(b)HT,(c)MT+HT
Benzinger和Hüttinger[3-5]证明了表面沉积反应是非均相反应,并与均相(热解)反应相互竞争。其中非均相反应有两种历程[6,7]:生长历程和形核历程。生长历程依靠基层表面的活性点化学吸附气相中以线型小分子和小分子芳香烃为主的活性组分,并在石墨烯层边缘生长,符合经典的Langmuir-Hinshelwood动力学方程。形核历程依靠基层表面物理吸附以多环芳烃(PAH)等大分子为主的组分形成新的石墨烯层,因此吸附平衡很重要,但不需要活性点的存在。Dong等[8]采用ICVI,沉积温度为1 100 ℃时以甲烷为原料气在较大压力范围内研究了热解炭微观结构的形成机理,提出了在生长阶段热解炭织构生成的Particle-filler模型。他们认为生成高织构热解炭的必要条件是气相中存在比例最优化的芳香烃C6(Particle)与直链烃是C2(filler),气相中存在过多的芳香化合物,或者过多的直链烃,都将导致中低织构热解炭的生成。对CVI过程来说,由于炭纤维预制体的比表面积AS/VR大,难以实现饱和吸附,气相组分大量消耗在反应表面大的PAH分子难以在孔内形成,因此热解炭沉积符合生长历程[7]。Vignoles和Delhaes等[9,10]采用ICVI和丙烷为前驱气体,认为以小分子线性烃类易沉积生成中织构热解炭而小分子芳香烃类易生成高织构热解炭,并且通过动力学分析,初步表明热解炭织构转变是属于双稳态过程,两个稳态之间存在迟滞转变区间。
基于Langmuir-Hinshelwood(L-H)理论,以气相组分中占主要的小分子线性烃类C2(Filler)和小分子芳香烃类C6(Particle)作为基体炭前驱体,以MT和HT热解炭作为两种不同亚稳相,假设在基体表面单分子前驱体沉积形成MT热解炭而P-F双分子反应形成HT热解炭,即以Particle-filler概念模型为指导对热解炭沉积过程进行动力学分析,研究气相组分浓度比、初始C2浓度以及沉积温度对热解炭沉积和织构形成过程的影响。
2模型及参数设定
2.1热解炭沉积和织构形成非均相表面反应动力学模型
考虑小分子线性烃类C2和小分子芳香烃类C6这两种主要的气相组分,将沉积成炭的过程按照L-H理论分为组分吸附、表面组分解吸附和表面组分脱氢3个过程。同时,基于P-F概念模型将MT和HT热解炭作为炭的两种亚稳相,以气相中占优的两种中间组分作为基体炭前驱体(线性小分子烃和小分子芳香烃),考虑基体表面单分子沉积形成MT热解炭和表面P-F双分子反应形成HT热解炭的过程。如图2所示,反应机理中总共包含7种组分和9个动力学常数,不考虑初始气相烃类原料气对表面反应的影响,即假设入口原料气在进入反应室并到达沉积表面时已经完全转化为线性小分子烃类C2和芳香小分子烃类C6,此时两者的比值R为0。
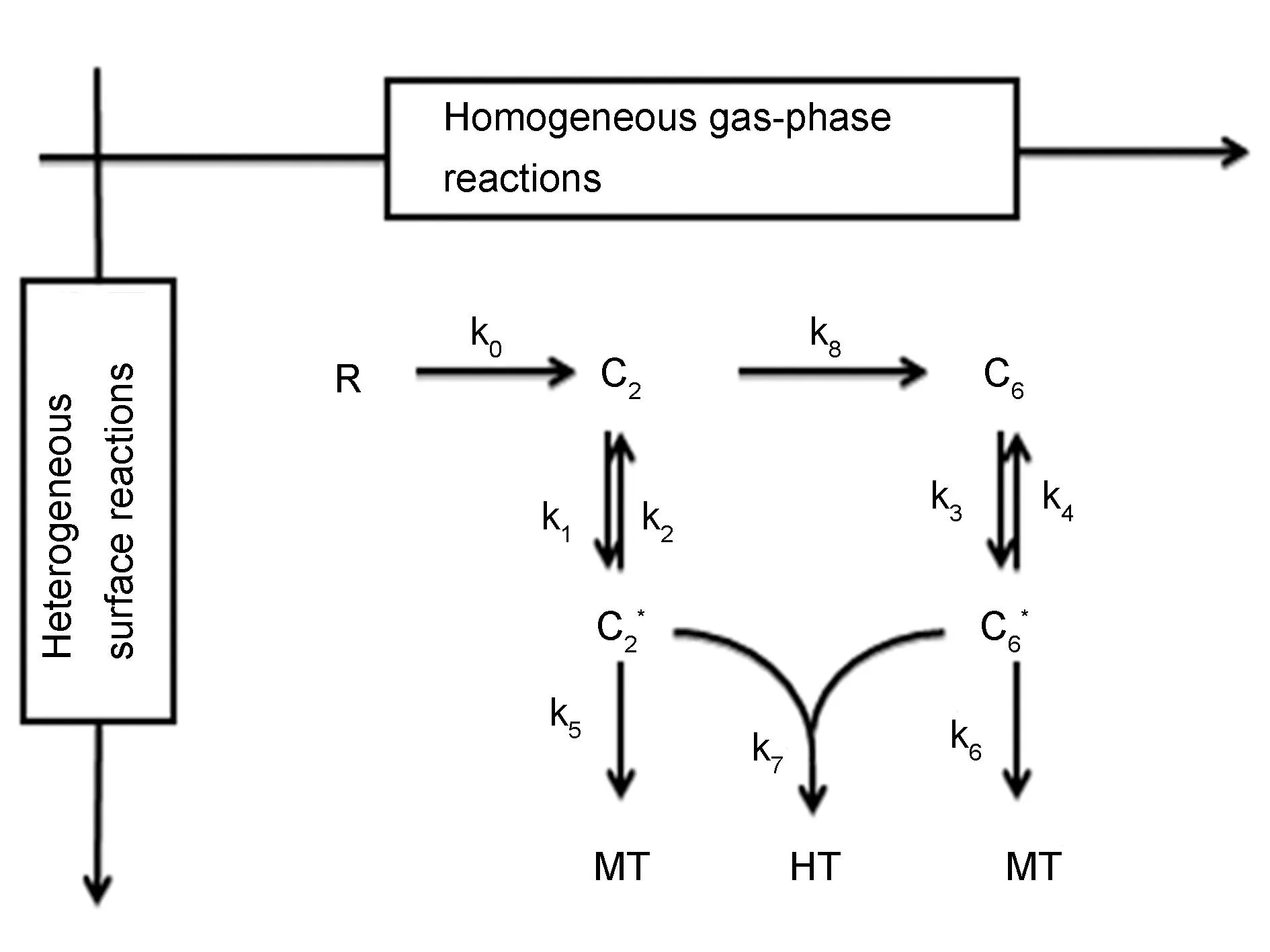
图 2 热解炭沉积和织构形成非均相表面反应动力学模型
探索稳态时基体近表面处不同的稳态气相组成对热解炭微观结构形成动力学的影响,因此不同气相组分间转化气相反应动力学不在讨论范围内,即k0=0,k8=0。同时,根据HACA机制乙炔在苯环上的加成反应可以得到完善的芳香烃结构,但在表面反应中整个域内单个或数个此类反应并不能决定热解炭的织构(图3),当表面上某两相邻域的边界分别存Aarmchair以及Zig-zag位就可能在两者之间会形成五元环以及七元环从而引起整个炭平面的上翘影响热解炭的织构。由此可知,整个域内的热解炭织构取决于整体的加成情况,即根据P-F概念模型只有F与P达到合适比例时,F才能完美填充P间的边缘形成完美的炭层,否则就会形成图3的非六元环。因此,仅以气相中占优的两种中间组分作为基体炭前驱体(线性小分子烃和小分子芳香烃)研究其在表面沉积过程中的宏观动力学特征。
基于反应机理(图2),具体得到如下多相化学反应:
(r1)
(r2)
(r3)
(r4)
(r5)
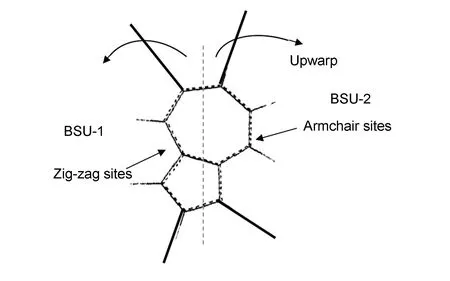
图 3 表面上两个基本结构单元之间
式中,S代沉积表面的活性位,C2H2*、C6H6*分别表示表面吸附的组分,主要是将P-F概念模型进行定量化,从而验证该模型的正确性。在该模型中将气相中所有的线性小分子烃以C2表示作为filler,将所有的大分子芳香烃以C6表示作为Particle,而对于各气相组分在基体表面发生吸附时占有的活性位目前还没有统一的定论,因此仅以含炭量来加以衡量,假设每个C2H2占有一个表面活性位,则每个C6H6占3个活性位,r1-r5均为基元反应,由此可建立表面吸附物浓度随时间变化的动力学方程为:
(e1)
(e2)
(e3)

当生成的热解炭织构和热解炭沉积速率不再随时间变化时,系统既达到稳态,此时表面[C2H2*]和[C6H6*]保持恒定,即式(e1)和(e2)满足下式:
(e4)
将上式e4代入(e1~e3)得:
(e5)
(e6)
(e7)
2.2非均相表面反应动力学模型设置
将表面组分覆盖率θ作为变量引入方程(e5)~(e7),并以C2H2的初始浓度[C2H2]0对系统进行无量纲化处理:
(e8)
(e9)
(e10)
式中:
a=[C2H2]/[C2H2]0,b=[C6H6]/[C2H2]0
(e11)
式中,反应速率常数ki与反应温度T的关系满足Arrhenius关系式:

(e12)
式中,Ai为指前因子,R为理想气体常数,ΔEi为反应的激活能。
文献[9]给出了沉积温度T为1 373 K时各反应速率常数ki,由(e12)可知只要确定相关反应的激活能就能计算出相指前因子等相关参数的值。取文献[11]的反应激活能作为相关反应的激活能值,见表1, 2。

表 1 文献中的相关表面反应激活能[11]
Note: C.(S)—active site; C(B)—deposited carbon; C6H6#—benzene.
3模拟结果与讨论
3.1气相组成对热解炭沉积的影响
Vignoles的热解炭沉积模型和Hüttinger的Particle-filler概念模型均认同气相组成对热解炭织构形成过程具有决定性影响。此处以气相中[C6]和[C2]比值R作为表征气相组分成熟度的主要变量,假设沉积表面处线性小分子烃C2归一化浓度a为1,沉积温度T为1 373 K,计算了随R值的变化(即随小分子芳香化合物C6归一化浓度的变化)热解炭沉积系统达到稳态时表面组分和热解炭织构的演变规律。求解(e8~e10)方程组在不同R值下的正实数解,如图4和5分别显示了C2与C6组分的表面覆盖率以及MT与HT热解炭的沉积速率随R的变化曲线,两组曲线都一致呈现“S”型,存在着上下两条实线分支和中间的虚线分支,为确定该“S”型曲线的三分支的状态,需要进行系统稳定性分析。所谓系统的稳定性,就是指给一个已经达到平衡状态的系统施加一个随机扰动,在扰动作用消失后经过一段过渡过程后能否回复到原来的平衡状态或足够准确地回复到原来的平衡状态的性能。若系统能恢复到原来的平衡状态,则称系统是稳定的;若扰动消失后系统无法恢复到原来的平衡状态,而是偏差越来越大,则系统不稳定[12]。

表 2 热解炭沉积和织构形成
Note:*activation energy from Table 1;#activation energy from gas-phase reaction[12]: A3+H=A3-4+H2and A3-4+C2H2=A3C2H2-4.
通过对系统进行线性稳定性分析[13]即可确定各分支的稳定性。将e3带入e1和e2,并进行无量纲化处理得到:
∂tx=k1a[C2H2]0(1-x-3y)-(k2+k5)x-k7[S]0xy
-k7[S]0xy(e13)

取x=x0+Δx,y=y0+Δy代入方程(e13)中,即分别给曲线上的某个x0和y0分别加上扰动Δx和Δy得到一个线性化扰动方程(e14):
(e14)
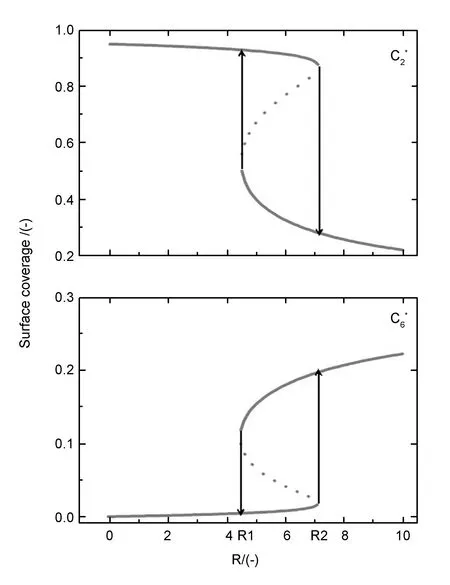
图 4 C2和C6表面覆盖率随R值的变化
式中:
a1=-(k1a[C2H2]0+k2+k5+k7[S]0y0
a3=-3k1a[C2H2]0+k7[S]0x0
此式的特征方程为:
γ2+c1γ+c2=0
(e15)
式中:
c1=k2+k4+k5+k6+k1a[C2H2]0+
按照Routh-Hurwitz判据[14,15],定态解x0和y0为稳定解的充要条件是:
(c1>0)∩(c2>0)
(e16)
若上式中有任一条件不满足,则系统的定态解失稳。显然c1中各项都大于零,所以针对图中各段只需计算c2是否满足(e16)即可。分别将图4和5的上中下三个分支曲线上点对应的(R,x0,y0)值代入c2表达式,易验证曲线的上下两分支满足(e16),而中间虚线分支不满足,因此,动力学上,上下两分支稳定而中间虚线分支是不稳定的,实验中该分支不存在,证明热解炭沉积过程是一个存在迟滞回线的双稳态过程。

图 5 HT和MT随R值的(a)沉积速率及其(b)沉积速率比
可见,热解炭沉积和织构形成过程是个含有迟滞回线的双稳态的动力学问题。当初始R较小时,随着R的增加C2的表面覆盖率沿着上分支缓慢减小,但此时表面主要还是由吸附的C2占据,结合图5可知,HT热解炭相对于MT热解炭沉积速率优势很小,可判定此阶段将导致整体的热解炭织构呈现MT织构特征;当R持续增加到R2时,C2的表面覆盖率将突变至下分支,之后沿着下分支变化,此时表面主要由大分子C6占据,主要沉积HT热解炭且沉积速率较大,可判定此阶段形成的热解炭将呈现HT织构的特征。当R值从较大值减小至R1时,HT沉积机制将会突变为MT沉积机制。在这过程中存在着如图上所示的回形迟滞区间,对于迟滞现象的产生,Vignoles认为,由基体表面的双分子反应引起的,而气相扩散与表面多相反应的相互作用则改变产生迟滞现象的参数区间[9]。
3.2线性小分子烃对热解炭沉积的影响
为分析线性小分子烃对热解炭沉积的影响,研究不同的线性小分子烃浓度条件下系统达到稳态时R值的变化对于热解炭沉积的影响。设定沉积温度T为1 373 K,线性小分子烃C2的归一化浓度a分别为0.1、0.5、1,通过求解(e8~e10)方程组,获得其正实数解,并对结果进行稳定性分析,见图6。

图 6 不同初始小分子浓度C2(归一化浓度)对于沉积反应的影响
从6图可以看出当线性小分子烃C2初始归一化浓度较小时,随R的增加,表面将由MT沉积机制急剧转变为HT沉积机制,且在沉积过程中不存在迟滞回线;随着初始线性小分子烃C2归一化浓度的逐渐增加,(e8~e10)方程组系统开始出现非稳态解,表面MT与HT热解炭沉积机制的区域间开始出现迟滞现象,且迟滞回线区间逐渐增加,而MT/HT热解炭沉积机制间的突变点也逐渐向右移动。
3.3沉积温度对于热解炭沉积的影响
假设初始线性小分子烃C2归一化浓度a为1,在不同沉积温度T为1 000、1 100、1 200 ℃条件下,通过求解(e8~e10)方程组,获取其正实数解,并对结果进行稳定性分析,见图7。图7与图6规律相似,即在沉积温度T较低时,随R的增加,表面各组分覆盖率会产生急剧变化,但并未出现迟滞的现象;随着沉积温度T的升高,表面MT与HT沉积机制的区域间开始出现迟滞且迟滞区间逐渐增加,而MT/HT沉积机制间的突变点也逐渐向左移动。即随着沉积温度的增加,体系由MT沉积机制突变为HT沉积机制时所需达到的R值越小。说明在一定条件下升高沉积温度T,可以使得系统由MT沉积机制转变为HT沉积机制。

图 7 不同温度下表面各组分的覆盖率变化
固定C2的初始归一化浓度a=1,在R分别为1、3、6时,求解(e7~e9),取不同沉积温度下的正实数解,并对结果进行稳定性分析。得到不同R值下表面吸附的C2和 C6组分的浓度随沉积温度的变化曲线(图8),在CVI反应过程中,气相烃类气体的热解以及热解产物在基体表面的吸附、脱附、沉积以及表面双分子反应本身都是吸热与放热的过程,因此即使在I-CVI过程中,体系内各处都会伴随温度的小范围波动,当R很小时,体系内温度的波动不会打破该处的反应平衡;只有当R大于某一临界值时,体系内温度的波动才有可能打破该处的反应平衡,如图7,对应R为3和6时的表面各组分覆盖率曲线,可以看出当体系处于C2下分支且远离右端点或处于上分支且远离左端点时(对应于C6上分支且远离右端点或下分支且远离左端点),体系内微小的温度波动不会打破该处的反应平衡。而当体系某处处于C2下分支的右端点(对应于C6上分支右端点)附近时,该处温度略微的向下波动将会导致该处由HT沉积机制突变为MT沉积机制;同样地,而当体系某处处于C2上分支的左端点(对应于C6下分支左端点)附近时,该处温度略微的向上波动将会导致该处由MT沉积机制突变为HT沉积机制。
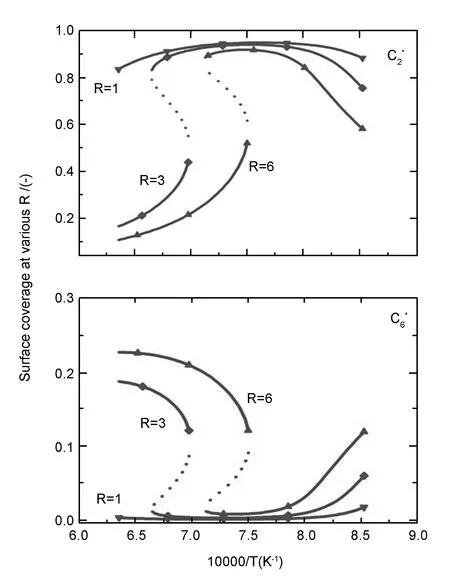
图 8 表面吸附C2和C6组分的浓度随温度的变化
3.4讨论
在CVI致密化过程,气相中中大小分子浓度比R,初始小分子浓度以及沉积温度T都对表面MT与HT热解炭的形成有很大的影响。对于前两种影响因素,可以通过原料气的选择以及初始浓度、预制体初始孔隙比表面积(A/V)、反应气体在入口处的滞留时间来进行控制。
在低压时,CVI系统中初始A/V相对较大,且随着反应的进行A/V逐渐变的更大,气相连续反应受阻,此时气相组分主要由小分子直链烃组成,即R较小,由图4和5可知此时表面主要由小分子C2占据,以MT热解炭沉积为主且沉积速率较小。此时若想将体系由MT形成机制转变为HT形成机制,由图7和8可知,提高体系的沉积温度T可以使得MT沉积机制向HT沉积机制转变,且R越小,需要的沉积温度T越高;
在中压时,甲烷分压的增加,体系中大分子芳香烃C6持续增加,此时基体表面不易达到饱和吸附,当体系达到稳定时,气相中生成的C6等于基体表面因反应消耗的C6,从而阻止了C6在气相中的持续长大。此时,R值较大,基体表面主要沉积HT热解炭且沉积速率较快;随着沉积的进行,A/V逐渐增加,气相中C6的消耗速率逐渐增加,从而使R逐渐减小,由图4可知如果R值降为R1,体系将会由HT形成机制突变为MT形成机制;由图7可知,若在此沉积过程中逐渐升高沉积温度T,体系由HT沉积机制转变为MT沉积机制的突变点R1将会逐渐减小,从而避免因为沉积过程中由于A/V比的不断增加导致系统由HT沉积机制向MT沉积机制转变。
在高压时,此时基体表面容易达到过饱和吸附,导致气相中烃类气体的不断长大,此时,气相主要由多环芳香烃与PAH组成,热解炭生成向形核机制转变,基体表面主要沉积MT热解炭。
此结论很好的解释了Rezinik等采用热壁反应器在1 100 ℃下甲烷的ICVI实验结果[16],即当甲烷分压为5 kPa时,纤维表面依次向外沉积了中织构与低织构炭:此时R较小,表面主要由小分子C2占据,随着沉积的进行,A/V逐渐增大,R持续减小,因此整个过程以MT热解炭沉积为主;当甲烷分压为10 kPa时,纤维表面沉积的是纯高织构炭:此时R较大,符合HT沉积机制,随着沉积的进行A/V逐渐减小,R持续减小但未降至R1以下,因此整个沉积过程都处于HT沉积机制;而当甲烷分压为20 kPa时,纤维表面先形成中织构炭后再形成高织构炭:初始沉积时基体表面已达到过饱和吸附,导致气相中烃类气体的不断长大,导致MT热解炭的沉积,随着基体表面不断地沉积A/V逐渐增大,使得整个反应系统脱离成核机制重新回归生长机制开始沉积HT热解炭。
Pauw在1 100 ℃,总压为100 kPa(10%甲烷,90%氩气)下研究了热壁反应器中滞留时间τ对于在硅表面沉积的热解炭织构演化的影响[17],见图9,当τ较小时主要沉积MT且沉积的炭颗粒平均直径较小,只有当τ大于某一临界值时才会沉积HT热解炭且沉积的炭颗粒平均直径较大(在该实验条件下,临界τ为1.4 s)。这主要是由于在τ较小时,气相组分以小分子直链烃为主,基体表面主要是小分子的沉积(MT沉积机制),所以沉积的炭颗粒平均直径较小;当τ较大时,气相组分不再以小分子为主,相当一部分已由大分子占据,此时表面主要发生的是双分子反应(HT沉积机制),此时沉积的炭颗粒平均直径较大。

图 9 在无氧条件下沉积1.5 h,Si表面沉积炭颗粒的
Hu等[6]利用热壁反应器研究了甲烷压强为25 kPa时,在不同滞留时间以及A/V值下董青石表面沉积的热解炭织构随沉积温度的变化。如图10所示,在较短的滞留时间(0.125 s)和高的A/V值条件下,热解炭气相沉积受化学生长模式控制,所得材料的织构在1 100~1 200 ℃存在最大值(OA最小值),说明该条件下对高织构的生成最优[18]。在此只关注温度对于热解炭沉积织构的影响,由图8可知,在沉积温度低于1 060 ℃左右时,不论R值多大,表面都是沉积MT热解炭;随着沉积温度的增加,R值大的体系将先于R值小的体系由MT沉积机制突变为HT沉积机制,这在图10中得到了很好的验证,即在A/V值为0.79 mm-1滞留时间为1 s时,体系由MT沉积突变为HT沉积的临界温度为1 040 ℃左右,而滞留时间为0.125 s时,临界温度为1 080 ℃度左右;同样在A/V值为3.2 mm-1也表现出了相似的趋势。由图7发现,随着沉积温度的升高,体系由MT沉积突变为HT沉积的所需达到的R值越小,说明在生长机制阶段沉积温度越高,体系越容易沉积HT热解炭,而在实验中我们发现在A/V值为0.79 mm-1滞留时间为1 s时,当沉积温度达到1 240 ℃时,体系将由HT沉积突变为MT沉积,与模拟结果不符合,这主要是由于在的模型中未考虑气相反应对于表面沉积的影响,随着沉积温度的升高,气相烃类气体的裂解速率逐渐大于气相反应中间产物在基体表面的吸附速率而导致基体表面的饱和吸附,从而使得气相组分逐渐长大,体系由生长机制转变为成核机制,从而导致由HT沉积机制转变为MT沉积机制。Guellali等研究得到如果给于充足的滞留时间让多环芳香烃分子内重排,形核机制也可以导致高织构热解炭的生成[19]。
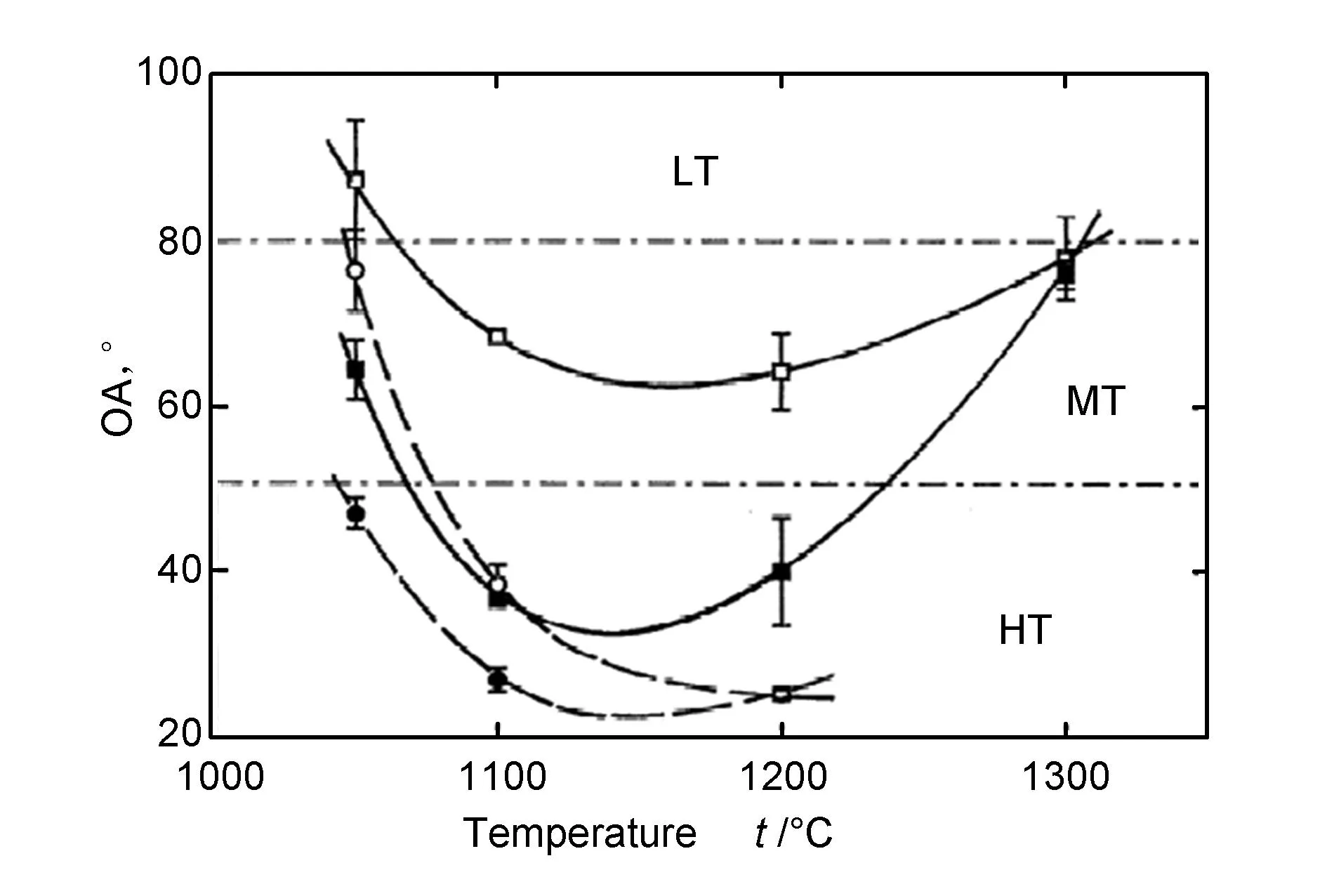
图 10 甲烷压强25 kPa、A/V值分别为0.79 mm-1(○/●)和
4结论
为理解CVI沉积过程中热解炭微观织构的突变现象,根据Langmuir-Hinshelwood理论并量化Particle-filler模型,建立了一个简化的热解炭沉积和织构形成模型,证实了在一定条件下热解炭的沉积是一个双稳态过程(分别是以MT和HT沉积机制主导的稳态过程)并存在一个迟滞区间,且此迟滞区间受气相组分浓度比,初始直链烃C2浓度以及沉积温度T的影响。量化的P-F模型能很好地解释热解炭结构的突变现象以及多种沉积条件下以甲烷为原料气的热解炭织构的演化过程。由于此模型将整个CVI过程中参与表面沉积的组分简化成了小分子直链烃C2类与大分子C6类并未考虑其它组分的沉积,也未考虑气相热解反应对于表面吸附组分的影响,因此,不能完全解释个别的工艺参数设置下CVI过程中热解炭沉积过程。
下一步将适当的增加参与表面沉积的气相组分,引入Monto Carlo方法研究热解炭的沉积过程,结合表面沉积机制研究整个CVI系统中热解炭沉积过程,从而模拟整个CVI过程中热解炭沉积演变过程。
参考文献
[1]Fitzer E, Manocha L M. Carbon Reinforcements and Carbon-carbon Composites[M]. Berlin, Heidelberg: Springer, 1998: 190-226.
[2]Beznik B, Guellali M, Gerthsen D, et al. Microstructure and mechanical properties of carbon/carbon composites with multilayered pyrocarbon matrix[J]. Mater Lett, 2002, 52: 14-19.
[3]Hüttinger K J. CVD in hot wall reactors-The interaction between homogeneous gas-phase and heterogeneous surface reactions[J]. Chem Vap Deposition, 1998, 4: 151-158.
[4]Benzinger W, Becker A, Hüttinger K J. Chemistry and kinetics of chemical vapor deposition of pyrocarbon: I. Fundamentals of kinetics and chemical reaction engineering[J]. Carbon, 1996, 34: 957-966.
[5]Benzinger W, Hüttinger K J. Chemical vapor infiltration of pyrocarbon: I. Some kinetic considerations[J]. Carbon, 1996, 34: 1465-1471.
[6]Hu Z, Hüttinger K J. Influence of pressure, temperature and surface area/volume ratio on the texture of pyrolytic carbon deposited from methane[J]. Carbon, 2003, 41: 749-758.
[7]Hu Z, Hüttinger K J. Mechanisms of carbon deposition-a kinetic approach[J]. Carbon, 2002, 40: 617-636.
[8]Dong G, Hüttinger K J. Consideration of reaction mechanisms leading to pyrolytic carbon of different textures[J]. Carbon, 2002, 40: 2515-2528.
[9]Vignoles G L, Baconneau O, Brauner C M. Interaction between gas diffusion and multistable heterogeneous chemical kinetics in C/C composite processing[J]. Fundamentals of gas-phase and surface chemistry of vapor phase deposition II/Process control, diagnostics and modeling in semiconductor manufacturing IV, 2001, 13: 237-244.
[10]Delhaes P. Attempts to chemical vapour infiltrate pyrocarbons: evidence for a spatial bistability[J]. Carbon, 2003, 41: 1093-1095.
[11]Lacroix R, Fournet R, Ziegler D I, et al. Kinetic modeling of surface reactions involved in CVI of pyrocarbon obtained by propane pyrolysis[J]. Carbon, 2010, 132-144.
[12]徐伟, 张中伟, 白瑞成, 等. 丙烷化学气相沉积均相热解反应动力学模拟[J]. 新型炭材料, 2014, 29: 67-77.
(Xu W, Zhang Z, Bai R, et al. Kinetic modeling of gas-phase reactions for CVD from propane[J]. New Carbon Mater, 2014, 29: 67-77.)
[13]胡寿松.自动控制原理[M]. 北京: 科学出版社, 2008: 1-632.
(Hu S. Principle of Automatic Control[M]. Beijing: Science Press, 2008: 1-632.)
[14]Lv L. Modulation effect of control parameter on chemical bistable system[J]. Journal of Shenyang power higher school, 2003, 3: 3-4.
[15]Xu D. Routh table and the stability of linear time-invariant system[J]. Chinese Science Bulletin, 1984, 8: 454-456.
[16]Reznik B, Gerthsen D, Zhang W, et al. Texture changes in the matrix of an infiltrated carbon fiber felt studied by polarized light microscopy and selected area electron diffraction[J]. Carbon, 2003, 41: 369-384.
[17]Pauw V D, Collin A, Send W, et al. Deposition rates during the early stages of pyrolytic carbon deposition in a hot-wall reactor and the development of texture[J]. Carbon, 2006, 44: 3091-3101.
[18]张伟刚. 化学气相沉积-从烃类气体到固体炭[M]. 北京:科学出版社, 2007: 12-128.
(Zhang W. Chemical Vapor Deposition from Hydrocarbon Gas to Solid Carbon[M]. Beijing: Science Press, 2007: 1-40.)
[19]Guellali M. Textures of pyrolytic carbon formed in the chemical vapor infiltration of capillaries[J]. Carbon, 2003, 41: 97-104.
Foundationitem: Doctoral Fund of Ministry of Education(20113108120019); National Natural Science Foundation of China(11202124); Aeronautical Science Foundation of China(2013ZF6001); Fund of Shanghai Municipality(13521101202).
Authorintroduction: HUANG Qing-bo, Master Student. E-mail: hqb_0222@shu.edu.cn
Simulation of the kinetics of pyrolytic carbon deposition in C/C composites
HUANG Qing-bo1,ZHANG Dan2,BAI Rui-cheng1,LI Ai-jun1,SUN Jin-liang1
(1.ResearchCenterforCompositeMaterials,ShanghaiUniversity,Shanghai200072,China)(2.ShanghaiInstituteofAppliedMathematicsandMechanics,ShanghaiUniversity,Shanghai200072,China)
Abstract:A multistep heterogeneous reaction kinetic model for pyrocarbon deposition is proposed to investigate the sharp and clear transition between the high-texture (HT) and medium-texture (MT) pyrocarbons in C/C composites synthesized by chemical vapor infiltration (CVI). The model is based on the Langmuir-Hinshelwood mechanism and a particle-filler conceptual model, which models both the pyrocarbon deposition and the texture formation. The model assumes that adsorption, desorption and dehydrogenation reactions are involved. Unimolecular dehydrogenation reactions of either light linear hydrocarbons as the source of fillers (F) or light aromatic species as the source of particles (P) result in the formation of MT pyrocarbon, while a bimolecular dehydrogenation reaction between P and F species leads to the formation of HT pyrocarbon. The relationship between the types of pyrocarbons and gas-phase compositions is simulated under steady state. It is found that MT and HT pyrocarbon formation are two dominant stable processes with a hysteresis interzone that is affected by gas composition, initial linear hydrocarbon concentration and deposition temperature. Simulated results account for the sharp and clear transition between MT and HT pyrocarbon, and agree well with most pyrocarbon evolution studies under various conditions during CVI with only a few exceptions that may be caused by simplification in constructing the model.
Keywords:Carbon/carbon composites; Pyrolytic carbon; Texture; Kinetics
文章编号:1007-8827(2016)02-0167-09
中图分类号:TQ342+.74
文献标识码:A
收稿日期:2016-01-05;修回日期:2016-04-02
基金项目:教育部博士点基金(20113108120019);国家自然科学基金(11202124); 航空科学基金(2013ZF6001); 上海市科委基金(13521101202).
通讯作者:李爱军,教授. E-mail:aijun.li@shu.edu.cn
作者简介:黄清波,硕士研究生. E-mail: hqb_0222@shu.edu.cn
Corresponding author:LI Ai-jun, Professor. E-mail: aijun.li@shu.edu.cn
