雷迪帕韦及其关键中间体的合成研究
2016-06-03赵聿秋孙光祥
赵聿秋+孙光祥
摘 要 乙醛酸甲酯与R(+)-α-甲基苄胺缩合、与环戊二烯通过aza-Diels–Alder 反应,产物经氢化、上Boc保护、水解、缩合得到苯并咪唑衍生物,继续与联硼酸频哪醇酯反应得到雷迪帕韦关键中间体:(1R,3S,4S)-3-[6-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1H-苯并咪唑-2-基]-2-氮杂双环[2.2.1]庚烷-2-羧酸叔丁酯,总收率40.6%(以乙醛酸甲酯计),关键中间体经过三步反应得到雷迪帕韦丙酮合物,纯度99.7%,ee值达100%。
关键词 雷迪帕韦 丙型肝炎治疗药物 关键中间体 合成
中图分类号:TQ463.55; R978.7 文献标识码:A 文章编号:1006-1533(2016)09-0065-06
An improved synthesis of ledipasvir and its key intermediate
ZHAO Yuqiu*, SUN Guangxiang
(Changzhou Pharmaceutical factory, Changzhou pharmaceutical company,
Shanghai Pharmaceuticals Holding Co. Ltd, Changzhou 213018, China)
ABSTRACT After the condensation of methyl aldehyde and D(+)-alpha-methylbenzylamine, followed by an aza-DielsAlder reaction with cyclopentadiene, a benzimidazole derivative compound was prepared via hydrogenation, Boc protection, hydrolysis and condensation, followed by reacting with coupling boric acid ester to obtain the key intermediate of (1R,3S,4S)-3-[6-(4,4,5,5-Tetra-methyl-1,3,2-dioxaborolan-2-yl)-1H-benzimidazol-2-yl]-2-azabicyclo[2.2.1]heptane-2-carboxylic acid 1,1-dimethylethyl ester. Ledipasvir could be prepared by using this key intermediate via three steps and its HPLC purity and ee value could reach to 99.9% and 100%, respectively.
KEY WORDS ledipasvir; anti-hepatitis C drugs; key intermediate; synthesis
雷迪帕韦(ledipasvir)化学名:N-[(2S)-1-[(6S)-6-[5-[9,9-二氟-7-[2-[(1S,2S, 4R)-3-[(2S)-2-(甲氧基羰基氨基) -3-甲基丁酰基] -3-氮杂双环[2.2.1]-2-庚基] -3H-苯并咪唑-5基] -2-芴基] -1H -2-咪唑基] -5-氮杂螺烷[2.4]-5-庚基] -3-甲基-1-氧代-2-丁基]氨基甲酸甲酯,是由吉利德科学公司开发,与该公司另一抗丙肝重磅产品Sovaldi(通用名:索非布韦)复方组合,商品名:Harvoni(复方索非布韦/雷迪帕韦),美国FDA于2014年10月10日批准其上市,同年11月21日获欧盟上市许可,是一种全口服,一天一次的片剂,用于治疗基因1型的丙型肝炎感染。作用机制:复方制剂中雷迪帕韦是一种丙肝病毒复制所需的HCV NS5A蛋白质抑制剂,索非布韦是HCV NS5B RNA-依赖RNA聚合酶抑制剂。Harvoni既可单药使用,也可以和其它口服制剂比如利巴韦林联合使用。
丙型病毒性肝炎是一种由丙型肝炎病毒(HCV)感染引起的病毒性肝炎。据世界卫生组织统计,全球HCV的感染率约为3%,共约1.8亿人。HCV感染主要包括免疫介导和HCV直接损伤两种方式,感染以后导致肝脏发炎以致肝功能下降甚至衰竭,病理表现以肝细胞坏死和淋巴细胞浸润为主。大多数的丙肝患者感染以后很久才发现肝损伤症状。感染20至30年有10%~20%的患者发展为肝硬化,1%~5%患者会因发生肝细胞癌(HCC)而死亡。而且肝硬化一旦出现失代偿情况,其生存率会急剧下降[1]。
Harvoni上市后凭其优异的治疗效果和较小的副作用,迅速得到医生和患者的认可,2015年销售额达到了惊人的138.6亿美元,显示出其巨大的市场潜力和在丙肝治疗领域无可替代的地位。
文献报道的雷迪帕韦合成路线主要有2条(图1):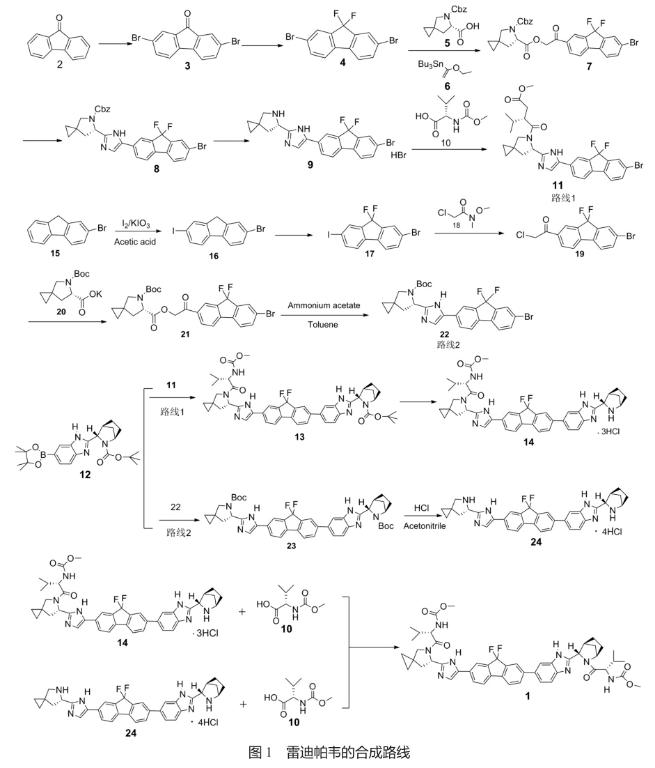
路线1采用9-芴酮(2)为原料,通过溴代反应、氟化反应得到偕二氟化化合物4,继续与三丁基(1-乙氧基乙烯)锡反应,再与5缩合得到7,7经重排环合、脱CBz保护得到9,继续与MOC-L-缬氨酸(10)缩合得到11。
路线2采用2-溴芴(15)为原料,通过碘代、氟化反应得到偕二氟化化合物17,17与2-氯-甲氧基-甲基乙酰胺(18)反应得到19,继续与20缩合、再进行重排环合得到22。
文献报道的2条雷迪帕韦(1)合成路线分别使用11和22同化合物12进行偶联缩合反应,再经过后续的脱保护、与MOC-L-缬氨酸(10)缩合反应得到目标产物1。
两条路线中都使用到关键中间体(1R,3S,4S)-3-[6-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1H-苯并咪唑-2-基]-2-氮杂双环[2.2.1]庚烷-2-羧酸叔丁酯(12), 12结构中含有3个手性中心,加之结构中的硼酸酯性质不够稳定,其质量状况直接影响到1的质量和制备收率,对1的产业化生产起着关键性的作用,所以对中间体12进行细致的合成研究,提高其收率和质量,使合成工艺适合工业化生产显得尤为重要。
文献[3-10]报道化合物12及其过程中间体的合成,以乙醛酸甲酯为原料,与R(+)-α-甲基苄胺缩合得到席夫碱化合物,席夫碱化合物与环戊二烯可以在DMF溶剂中通过aza-Diels–Alder 反应[4],产物通过柱层析纯化得到(1S,3S,4R)-2-((1R)-1-苯基乙基)-2-氮杂双环[2.2.1]庚-5-烯-3-羧酸甲酯,收率89%,外型∶内型(69:31),工艺不适合工业化放大生产,内型杂质含量高,不利于后续反应进行;席夫碱化合物与环戊二烯也可以在三氟化硼乙醚和三氟乙酸催化下通过aza- Diels–Alder 反应得到29粗品,直接经钯碳催化的氢化还原[5],收率仅32.6%,不适合工业化放大生产的成本控制,且使大量的物料浪费,增加环保压力;中间体29经氢化还原、上BOC保护、水解,再与4-溴邻苯二胺缩合得到一对同分异构体混合物,该混合物通过加热闭环反应得到苯并咪唑衍生物,继续与联硼酸频哪醇酯反应得到(1R,3S,4S)-3-[6-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1H-苯并咪唑-2-基]-2-氮杂双环[2.2.1]庚烷-2-羧酸叔丁酯(12)[3],文献报道12反应液直接用于下一步反应[3],由于12反应液中杂质多,导致下一步反应副产物多,部分杂质难以去除,最终得到的1成品质量难以符合ICH指南的要求。
文献报道的合成12的工艺,普遍存在反应收率低、反应过程中内型异构体杂质含量高、部分中间体的纯化需要柱层析,部分杂质难以去除等不利因素[3-7],不适合工业化的生产需要。
针对以上不足,本研究对12的合成工艺进行改进(图2),具体体现在:制备(1S,3S,4R)-2-((1R)-1-苯基乙基) -2-氮杂双环[2.2.1]庚-5-烯-3-羧酸甲酯(29),文献报道的两种方法,一种是通过柱层析纯化得到一个内型和外型的混合物,另一种收率过低,本研究采用三氟化硼乙醚和三氟乙酸催化,适当降低反应温度至-65 ~-75 ℃,减少反应过程中的副产物杂质,改进后处理操作,粗品采用石油醚精制,得到了纯化的中间体,收率60.8%,对后续反应的收率和质量控制带来了明显的提高(文献收率32.6%[5])。文献[3]报道的12制备过程是粗品12直接进行下一步反应,我们研究发现,粗品中的杂质会参与后续反应,形成杂质传递,导致最终1的质量很难符合ICH指南的要求,文献[2]通过柱层析纯化得到12,本研究通过适当减少反应时间,TLC监控反应原料转化完全,减少高温反应过程中产物本身的部分分解,后处理增加洗涤、萃取、干燥、活性炭脱色过程,所得粗品通过正庚烷打浆,分离得到高质量的12,收率提高到93.8%(文献收率85%[2]),实现了含贵金属催化剂的溶液的分离和回收,大大节约了原料成本。优化后的工艺操作简便、质量提高,制备中间体12的总收率:40.6%(文献[3-7]总收率:15.0%),适合工业化生产。
通过我们的研究,解决了文献工艺中不适合工业化生产的柱层析纯化操作,显著提高了收率,中间体12的总收率由文献[3-7]报道的总收率15.0%,提高到40.6%,优化后的工艺操作简便,适合工业化生产,同时关键中间体12质量提高,有利于1成品的质量控制和制备收率的提高。对文献报道的1合成路线进行评估,选择了图3所示的路线合成1成品,通过对后处理及精制条件的优化,所得产品纯度达到99.7%(HPLC面积归一化法),ee值100%,符合ICH指南的质量要求。工艺简便,适合工业化生产。
由于工艺中的aza-Diels–Alder反应过程中产品构型的选择性在现有文献报道和我们研究使用的催化剂条件下,需要低温条件下反应(-65℃以下),才能得到理想的97:3的外型:内型选择性,虽然低温反应在工业化生产上已经是个成熟的工艺,但生产过程中需使用液氮冷却,存在能耗高、有一定的安全隐患的问题。下一步研究的重点是找到一个创新性的、高效的、耐受度高的催化条件,能够在车间易于实现的-5℃以上温度条件下,也能达到97:3的外型:内型的选择性,使工艺过程更加低碳、安全。
1 实验部分
1.1 (1S,3S,4R)-2-((1R)-1-苯基乙基)-2-氮杂双环[2.2.1]庚-5-烯-3-羧酸甲酯(29)
向5 L反应中加入二氯甲烷(2 L)、4A分子筛(200 g)和乙醛酸甲酯(197.7 g,2.25 mol),搅拌10 min,室温下滴加R(+)-α-甲基苄胺(272.1 g,2.25 mol),保持室温搅拌过夜。过滤,母液搅拌冷却至-65~-75 ℃,控制温度低于-65 ℃依次滴加三氟乙酸(245.67 g,2.36 mol)和三氟化硼乙醚(327.7 g,2.36 mol),缓慢滴加环戊二烯(155.8 g,2.36 mol),保持-65~-75 ℃反应2 h,缓慢升温至室温反应过夜,加入1.5 L水,碳酸氢钠水溶液调节pH值8~9,分层,有机相浓缩至干,加乙酸乙酯(2 L)溶解,加入3 mol/L盐酸(1 L),搅拌、分取水相用碳酸氢钠调节pH值至8~9,石油醚(1 L×3)提取,无水硫酸钠(100 g)干燥,过滤,母液减压浓缩至剩余体积800 ml左右,搅拌冷却至0~5 ℃析晶过夜,过滤得352.1 g白色固体29,收率:60.8%。1HNMR (400 MHz,DMSO) δ7.19~7.11(m,5H),6.42(t,1H),6.32(m,1H),4.17(d,J=1.3 Hz,1H),3.21(s,3H),2.95(m,1H),2.82(d,J=1.3 Hz,1H),2.00(s,1H),1.94(m,1H),1.24(m,4H)。
1.2 (1R,3S,4S)- 2-氮杂双环[2.2.1]庚烷-3-羧酸甲酯(30)
向5 L四口反应瓶中加入无水乙醇(2.5 L)、29(257 g,1.0 mol)和5% Pd/C(12.8 g),氮气和氢气各置换3次,常压氢化反应12 h,过滤、钯碳回收套用,母液转移至5 L氢化反应釜,加入5% Pd/C(25.7 g),氮气和氢气各置换3次,10 bar压力下室温反应24 h,HPLC监控显示反应转化完全,过滤,母液45 ℃下减压浓缩至干,得油状物(159.7 g),直接用于下步反应。
1.3 (1R,3S,4S)-N-叔丁氧羰基-2-氮杂双环[2.2.1]庚烷-3-羧酸(32)
5 L四口烧瓶中加入30(159.7 g)的四氢呋喃(1.6 L)溶液,搅拌加入20%碳酸氢钠溶液(1.6 L),搅拌,室温滴加二碳酸二叔丁酯(BOC酸酐),滴毕,搅拌反应6 h,反应完毕,40 ℃下减压浓缩四氢呋喃至基本无液体蒸出,加入乙酸乙酯(1.5 L)和水(750 ml),搅拌分层,分取有机相用无水硫酸钠干燥(100 g),45 ℃下减压浓缩至干得油状物。
向上述油状物中加入1.5 L四氢呋喃,搅拌溶清,控制室温条件,滴加15%氢氧化钠溶液(1.8 L),保持25~30℃反应24 h,反应完毕,40 ℃下减压浓缩四氢呋喃至基本无液体蒸出,水层用石油醚(500 ml×3)提取,水相用3 mol/L盐酸(约2.3 L)调节pH值至4~5,加入乙酸乙酯(1 L)搅拌30 min,分层,水层用乙酸乙酯(800 ml)提取,合并有机相,饱和氯化钠溶液(800 ml)洗涤,无水硫酸钠(100 g)干燥,过滤,母液浓缩至干得固体粗品,加入500 ml正庚烷打浆3 h,过滤,30 ℃下真空干燥得白色固体197.8 g,收率:82.0%(以化合物29投料量计算)。1HNMR (400 MHz,DMSO) δ12.5(s,1H),4.07(d,J=29.5 Hz,1H),3.58(d,J=4.3 Hz,1H),2.57(m,1H),1.71~1.49(m,5H),1.40,1.36(s,9H),1.19(m,1H)。
1.4 (1R,3S,4S)-3-(6-溴-1H-苯并咪唑-2-基)-2-氮杂双环[2.2.1]庚烷-2-羧酸叔丁酯(36)
3 L四口烧瓶中加入32(150 g,0.62 mol)、1 L二氯甲烷,搅拌溶解,冷却至-5~0 ℃,依次投入HOBT(114.5 g,0.75 mol)、EDCI(143.4 g,0.75 mol),搅拌反应3 h,加入4-溴-邻苯二胺(116.0 g,0.62 mol),滴加N-甲基吗啉(188.6 g,1.86 mol),滴毕缓慢升温至20~25 ℃,搅拌反应过夜。加入饱和碳酸氢钠溶液(800 ml),搅拌,分层,分取有机相,水相用二氯甲烷提取(200 ml×2),合并有机相,无水硫酸钠(100 g)干燥,过滤,母液减压浓缩至干,油状物直接用于下步反应。
向上述油状物中加入乙酸(225 ml)、甲基叔丁基醚(1.5 L)搅拌加热至回流,保持回流反应18 h,降温至0~5 ℃,滴加15%氢氧化钾溶液(约1 400 g),调节pH值13~14,分层,水层用甲基叔丁基醚(200 ml×2)提取,合并有机相,饱和氯化钠水溶液(500 ml)洗涤,活性炭(20 g)脱色,过滤,母液减压浓缩至干,油状物用异丙醚(800 ml)精制,得白色固体(212 g),收率86.9%。1HNMR (400 MHz,DMSO) δ12.48~12.29(m,1H),7.71(s,0.5H),7.57(t,0.5H),7.48(d,J=8.5 Hz,0.5H),7.38(m,0.5H),7.27~7.22(m,1H),4.42(d,J=13.5 Hz,1H),4.19(d,J=37.2 Hz,1H),2.48(m,1H),1.94(d,J=10.0 Hz,1H),1.71~1.61(m,4H),1.39,1.23(s,9H),1.30(m,1H)。
1.5 (1R,3S,4S)-3-[6-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1H-苯并咪唑-2-基]-2-氮杂双环[2.2.1]庚烷-2-羧酸叔丁酯(12)
向3 L四口烧瓶中加入36(200 g,0.51 mol)、联硼酸频哪醇酯(155.4 g,0.61 mol)、丙酸钾(171.6 g,1.53 mol)、双(二叔丁基苯基膦)二氯化钯(11.5 g,0.018 mol)和醋酸异丙酯(1.5 L),搅拌加热至回流,保持回流2 h,TLC(石油醚-乙酸乙酯=1∶1)显示原料36转化完全,冷却至40~45 ℃,减压浓缩除醋酸异丙酯,加入二氯甲烷(1.5 L)和水(600 ml),搅拌溶解,分层,分取有机相用饱和氯化钠水溶液(300 ml)洗涤,有机相用无水硫酸钠(50 g)干燥,抽滤,母液减压浓缩至干,加入正庚烷(2 L)搅拌打浆6 h,过滤,少量正庚烷漂洗,45 ℃下真空干燥,得类白色固体(210.2 g),收率93.8%。1HNMR (400 MHz,DMSO) δ12.31(s,1H),7.77(s,1H),7.44(m,2H),4.45(d,J=10.0 Hz,1H),4.21(d,J=39.2 Hz,1H),2.61(m,1H),1.93(m,1H),1.69~1.59(m,4H),1.42(s,4H),1.33(m,13H),1.22(m,1H),1.13(m,4H)。
1.6 (6S)-6-[5-[7-[2-(1R,3S,4S)-2-氮杂双环[2.2.1]庚烷-3-基-1H-苯并咪唑-6-基]-9,9-二氟-9H-芴-2-基]-1H-咪唑-2-基]-5-氮杂螺[2.4]庚烷四盐酸盐(24)
向1 L反应瓶中加入22(27.1 g,0.05 mol)、12(22.0 g,0.05 mol)、碳酸钾(20.7 g,0.15 mol)、DMF(400 ml)、水(80 ml),氮气保护下加入四(三苯基磷)钯(5.78 g,0.015 mol),搅拌加热至75~80 ℃,保温反应12 h,冷却反应液至室温,加入醋酸异丙酯(250 ml)和750 ml水,搅拌分层,分取有机层,减压浓缩至干得固体粗品,粗品直接用于下一步反应。
向上述粗品中加入乙腈(150 ml)、4 mol/L盐酸(150 ml),搅拌升温至60~65 ℃,反应3 h,反应结束冷却至室温,加入乙腈(500 ml),室温搅拌析晶3 h,抽滤,50 ℃下鼓风干燥,得类白色固体(31.6 g),收率87.8%。MS(m/z): 575.273 4[M+H]+,1HNMR (500 MHz,DMSO)δ10.93(br,1H),10.45(br,1H),8.43(s,1H),8.39(s,1H),8.25(d,J=8.0 Hz,1H),8.07(m,2H),8.02(d,J=8.0 Hz,1H),7.99(d,J=8.5 Hz,1H),7.96 (d,J=8.0 Hz,1H),7.86(s,1H),7.81(d,J=8.5 Hz,1H),6.37(br,6H),5.39(t,1H),4.97(s,1H),4.26(s,1H),3.73(d,J=11.0 Hz,1H),3.31(s,1H),3.20(J=11.0 Hz,1H),2.94(m,1H),2.31(m,1H),2.18(m,1H),2.12(m,1H),1.91(m,1H),1.81(m,2H),1.75(m,1H),0.90(m,2H),0.76(m,2H)。
1.7 雷迪帕韦(1)
将MOC-L-缬氨酸(20 g,0.11 mol)、EDCI(21.1 g,0.11 mol)和二甲基甲酰胺(300 ml)依次加入至1 L反应瓶中,室温搅拌反应30 min,加入24(25 g,0.035 mol)和N-甲基吗啉(25 ml),加毕保持室温反应6 h,反应完毕,加入乙酸乙酯(200 ml)和水(150 ml),搅拌分层,水相用乙酸乙酯(200 ml)提取,合并有机相,分别用饱和碳酸氢钠(150 ml)、饱和食盐水(150 ml)洗涤,无水硫酸钠(30 g)干燥,抽滤,母液浓缩至干,加入丙酮(250 ml),加热溶解,搅拌冷却至10~15 ℃析晶过夜,过滤、滤饼用丙酮(250 ml)室温打浆3 h,过滤,35~40 ℃下真空干燥得类白色固体(23.5 g),收率76.1%;纯度99.7%(HPLC面积归一化法),ee值100%(手性色谱HPLC面积归一化法)。 MS(m/z): 889.420 2[M+H]+, 1HNMR (500 MHz,DMSO) δ12.19(d,J=3.0 Hz,1H),11.86(s,1H),8.12(s,1H),7.99(m,2H),7.87(m, 2H),7.82(m,2H),7.72(s,1H),7.67(m,1H),7.54(m,1H),7.29(d,J=8.0 Hz,1H),7.19(d,J=8.5 Hz,1H),5.25(m,1H),4.72(d,J=7.0 Hz,1H),4.57(s,1H),4.21(m,1H),4.06(m,1H),3.86(d,J=10.0 Hz,1H),3.75(d,J=10.0 Hz,1H),3.58(m,6H),2.70(s,1H),2.44(m,1H),2.24(m,1H),2.12(m,1H),2.09(s,丙酮甲基氢),2.04(m,2H)1.95(m, 1H),1.95(m,1H),1.78(m,2H),1.57(m,1H),1.47(m,1H),0.95~0.92(m,12H),0.73(m,1H),0.57(m,3H)。
参考文献
[1] 吉利德Harvoni获批, 抗丙肝新金标全口服方案出炉[EB/OL]. (2014-10-11)[2016-04-18]. http://www.sinohealth. com/2014/1011/10748.shtml.
[2] Ray AS, Watkins WJ, Link JO, et al. Methods for treating HCV: WO 2013040492[P]. 2013-03-21.
[3] Scott RW, Wang F, Shi B, et al. Solid forms of an antiviral compound: US 8,969,588 [P]. 2015-03-03.
[4] Bailey PD, Brown GR, Korber P, et al. Asymmetric synthesis of pipecolic acid derivatives using the aza-Diels-Alder reaction[J]. Tetrahedron Asymmetry, 1991, 2(12): 1263–1282.
[5] Tararov VI, Kadyrov R, Kadyrova Z, et al. An improved synthesis of enantiopure 2-azabicyclo[2.2.1]heptane-3-carboxylic acid [J]. Tetrahedron Asymmetry, 2002, 13(1): 25–28.
[6] Link JO, Taylor JG, Xu L, et al. Discovery of ledipasvir(GS-5885): a potent, once-daily oral NS5A inhibitor for the treatment of hepatitis C virus infection [J]. J Med Chem, 2014, 57(5): 2033-2046.
[7] Hashimoto N, Yasuda H, Hayashi M, et al. Aza-Diels?Alder reaction of methyl 2-[(r)-1-phenylethyl]iminoethanoate with cyclopentadiene using practical and environmentally friendly biphasic solvent system[J]. Org Process Res Dev, 2005, 9(1): 105-109.
[8] Bailey PD, Wilson RD, Brown GR. Enantio- and diastereoselective synthesis of pipecolic acid derivatives using the azaDiels–Alder reaction of imines with dienes[J]. J Chem Soc, Perkin Trans 1, 1991(5): 1337-1340.
[9] Dyatkin AB, Hoekstra WJ, Hlasta DJ, et al. Bridged bicyclic vasopressin receptor antagonists with V2-selective or dual V1a/ V2 activity[J]. Bioorg Med Chem Lett, 2002, 12(21): 3081-3084.
[10] Limburg DC, Thomas BE IV, Li JH, et al. Synthesis and evaluation of chiral bicyclic proline FKBP12 ligands[J]. Bioorg Med Chem Lett, 2003, 13(21): 3867-3870.
