纤锌矿Be1-xMgxO合金的电子结构和相特性研究
2016-05-25郑树文陈振世
郑树文,张 涛,张 力,陈振世
(华南师范大学 光电子材料与技术研究所,广东省光电功能材料与器件工程技术研究中心, 广州 510631)
纤锌矿Be1-xMgxO合金的电子结构和相特性研究
郑树文,张涛,张力,陈振世
(华南师范大学 光电子材料与技术研究所,广东省光电功能材料与器件工程技术研究中心, 广州 510631)
摘要:采用第一性原理的平面波超软赝势方法,对纤锌矿Be1-xMgxO合金的电子结构和相特性进行研究。结果显示,纤锌矿Be1-xMgxO的能隙由价带顶O2p态和导带底Mg3s态共同决定。随着Mg组分的增大,Be1-xMgxO合金的能隙逐渐变小,合金离子性在增强。Be—O和Mg—O的平均键长差距大致使Be1-xMgxO合金内部产生明显的晶格振动效应,而无序相Be1-xMgxO合金的形成能比有序相要低,容易使纤锌矿Be1-xMgxO合金内部产生无序结构,因此制备纤锌矿Be1-xMgxO合金时要适当提高实验温度。
关键词:第一性原理;电子结构;相特性;Be1-xMgxO
0引言
最近几年, 短波长光电器件和相关材料受到人们的广泛关注。 由于Ⅱ-Ⅵ族宽隙带材料(ZnO、MgO和BeO)在短波长方面具有优秀的物理和化学特性, 可望在紫外光电器件、太阳能电池、探测器、压电元件等领域发挥潜在应用价值而越来越受到研究者的重视[1]。 目前, ZnMgO和ZnBeO合金材料已经被业界进行了深入的研究,发现当ZnO中掺入Mg时能形成 MgxZn1-xO合金,但是常温下ZnO和MgO的相结构不同, 所以往ZnO掺入Mg时容易产生相问题[2]。 Ohtomo等[3]指出,当Mg组分高于0.36时,MgxZn1-xO合金就出现相分离, 由纤锌矿结构转变成岩盐矿结构,这样MgxZn1-xO合金的能隙调节范围受到制约。为此,Ryu等[4-5]提出用BexZn1-xO合金代替MgxZn1-xO合金的思想,通过HBD方法在c-Al2O3基板上制备出全Be组分的BexZn1-xO合金,对应的能隙范围从3.37~10.6 eV,拓宽了ZnO器件的能隙范围。
对于Be1-xMgxO合金的研究,目前文献报道的不多, Shi等[6]研究了闪锌矿Be1-xMgxO合金的能隙和弯曲系数随Mg掺杂组分的变化关系,文献[7]分析了Mg掺杂对纤锌矿和岩盐矿Be1-xMgxO合金的结构参数和能带特性的影响,指出纤锌矿和岩盐矿结构的Be1-xMgxO合金的能隙都为直接能隙,纤锌矿Be1-xMgxO合金能隙值在6.862~10.6 eV之间,而岩盐矿Be1-xMgxO合金的能隙值在7.8~11.24 eV之间,同时发现当Mg组分高于0.89时Be1-xMgxO合金的纤锌矿相向岩盐矿相转变,但文中没有对Be1-xMgxO合金的能带来源和相结构问题(如材料的有序和无序相)作分析。为了更深入理解Be1-xMgxO合金的物理特性,就需要对Be1-xMgxO合金的电子结构和相特性作更多研究。
第一性原理的研究方法已经广泛应用到材料物性的分析工作中[6-11]。例如,靳锡联等[10]采用平面波赝势理论方法研究了Mg掺杂对纤锌矿ZnO合金的电子结构影响,史力斌等[11]利用密度泛函理论对纤锌矿BexZn1-xO合金的电子结构和光学特性进行了理论分析,它们的理论研究结果与实验相一致。为此,本文采用第一性原理的平面波超赝势方法,对纤锌矿Be1-xMgxO合金的电子结构和相特性作深入研究,为下一步开展纤锌矿Be1-xMgxO合金的实验研究和相关工作提供指导。
1模型构建与计算方法
本文计算的纤锌矿Be1-xMgxO合金结构,是通过在纤锌矿BeO(2×2×1)超原胞中掺入不同Mg组分获得。 这样超原胞体系共含有16个原子,其中O原子个数为8个, Be和Mg原子个数共为8个。 当纤锌矿BeO超原胞中的2个Be原子被Mg原子取代时,就形成了纤锌矿Be0.75Mg0.25O三元合金。通过改变纤锌矿BeO超原胞中Mg原子取代Be原子的个数,得到Mg掺杂组分分别为0,0.25,0.5,0.75和1的纤锌矿Be1-xMgxO合金。注意的是,文中构建的计算模型是根据超原胞总能量最低值来决定纤锌矿Be1-xMgxO合金的稳定相,模型构建的初始晶格参数参考实验值[12-13]。
本文中计算采用的软件是基于密度泛函理论(DFT)[14]应用平面波赝势方法的CASTEP软件包[15]。 计算的电子间交换相关能用广义梯度近似(GGA)的PW91[16]泛函来描述,并用超软赝势[17]来描述离子实与价电子之间的相互作用势,选取Be、Mg和O原子的电子组态分别为Be2s2、Mg2p63s2和O2s22p4。为了得到高精度的计算结果,计算中的平面波截断能Ecut设为650 eV,Monkhorst-Pack[18]型的K网格点设为4×4×5。计算前先在不固定任何参数下进行几何优化,能量、原子间相互作用力、晶体内应力和原子最大位移的收敛精度分别设为5.0×10-6eV/atom、0.1 eV/nm、0.02 GPa和5.0×10-5nm。几何优化完成的标志是上述4个参数均达到和优于收敛精度。经优化计算得到的纤锌矿BeO和MgO结构参数和能隙值见表1所示。
表1纤锌矿BeO和MgO的晶格常数a、c和能隙Eg计算值与文献实验值(或理论值)对比
Table 1 Calculated structural parameters and energy bandgap of wurtzite(w)-BeO and w-MgO, comparison to experimental and other theoretical results

a/nmc/nmuEg/eV文献BeO0.27500.27180.27410.27140.26930.27750.44670.44080.44540.44130.43700.43850.3760.3770.3857.0810.67.2937.4447.00本文实验[12-13]理论[2]理论[19]理论[20]理论[21]MgO0.33440.33220.33060.51610.51360.51020.3920.39160.3873.3333.33.452本文理论[22]理论[23]
由表1数值知,纤锌矿BeO晶格常数比文献实验值[12-13]略大,但误差<1.2%,并与前人的理论值[2,19-21]接近。 优化后的纤锌矿MgO晶格常数也跟相关文献[22-23]的报道结果相符。 对于优化后的纤锌矿BeO能隙值,与实验文献[12-13]存在偏差,这是由于GGA近似下的DFT理论在处理材料带隙问题时没有考虑到研究体系的激发态情况,造成激发态电子间的关联效应被低估,从而引起理论值比实验值偏小的共性问题[24],但本文的纤锌矿BeO和MgO能隙值与其它文献理论值[20,22-23]相一致,所以说本文采用的计算方法和计算参数是可信的。
2结果与讨论
2.1纤锌矿Be1-xMgxO合金的总态密度和分波态密度
图1为纤锌矿Be1-xMgxO(x=0,0.25,0.5,0.75和1)合金的总态密度图(TDOS)。
图1的费米能级EF定义为能量零点。由图1可知,BeO的电子态密度主要分布在3个区域,即上价带(-6.4~0 eV)、下价带(-18.9~-15.3 eV)和导带(7.08~14.6 eV)。当Mg掺入纤锌矿BeO材料形成Be1-xMgxO合金时,Be1-xMgxO的电子态密度分布发生明显变化,在-38 eV附近产生一个电子占据态高峰,同时上价带、下价带和导带区域的电子态密度分布都发生移动,比如上价带朝价带顶方向压缩,其宽度由BeO的6.92 eV变成MgO的3.886 eV,与之对应的电子占据数分别为48.16 e和48.23 e。下价带的态密度分布位置也随Mg组分的增大朝价带顶方向移动,局域性进一步增强。对于导带受Mg掺杂组分增大的影响,主要体现在导带底不断朝费米能级方向移动,造成Be1-xMgxO合金的能隙逐渐变窄,同时导带的电子态分布更弥漫。
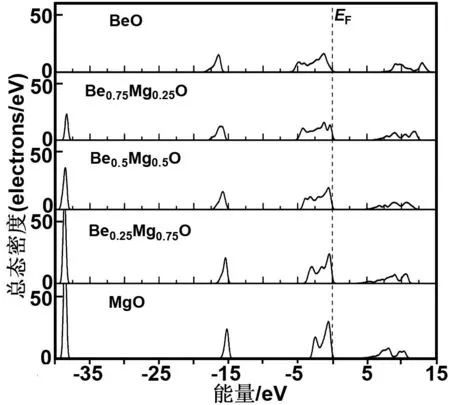
图1 纤锌矿Be1-xMgxO合金的总态密度
Fig 1 Total DOS of w-Be1-xMgxO, the Fermi level (EF) indicated by a vertical dashed line is set to be zero
为了清晰Be1-xMgxO合金总电子态密度对应的电子轨道分布情况,图2给出了纤锌矿Be1-xMgxO(x=0,0.5和1)合金的分波态密度图(由于-38 eV附近的电子态密度对材料性质影响小,故不绘于图2中)。由图2(a)知,纤锌矿BeO上价带的态密度主要由O2p态轨道占据,还包括少量的Be2s2p态。导带主要由Be2p态和少量Be2s和O2s态占据,而下价带主要由O2s态占据。由于下价带距离上价带顶较远,所以对BeO能隙的影响不大。 对于图2(b)的Be0.5Mg0.5O合金,上价带主要由O2p态轨道占据,但还包括少量的Be2s2p态和Mg3s态,由于O2p态占据了价带顶位置,所以其决定了价带顶的性质。导带主要由Be2p和Mg2p态占据,还包括少量的Be2s态和Mg3s态,但导带底位置由Mg3s态占据,所以Be0.5Mg0.5O合金的能隙性质由价带顶的O2p态和导带底的Mg3s态共同决定。对于Be0.5Mg0.5O合金的下价带态密度分布主要由O2s态和Be2s2p态占据,并表现出较强局域性。对于图2(c)的纤锌矿MgO态密度,其上价带主要由O2p态占据并决定了价带顶位置,而导带主要由Mg2p3s态占据,虽然Mg3s态占据数仅为3.09 e,不及Mg2p态占据数的1/5,但占据导带底位置的为Mg3s态,这里纤锌矿MgO的分波态密度分布结果与靳锡联[10]和李巧芬等[25]的理论计算结果相一致。
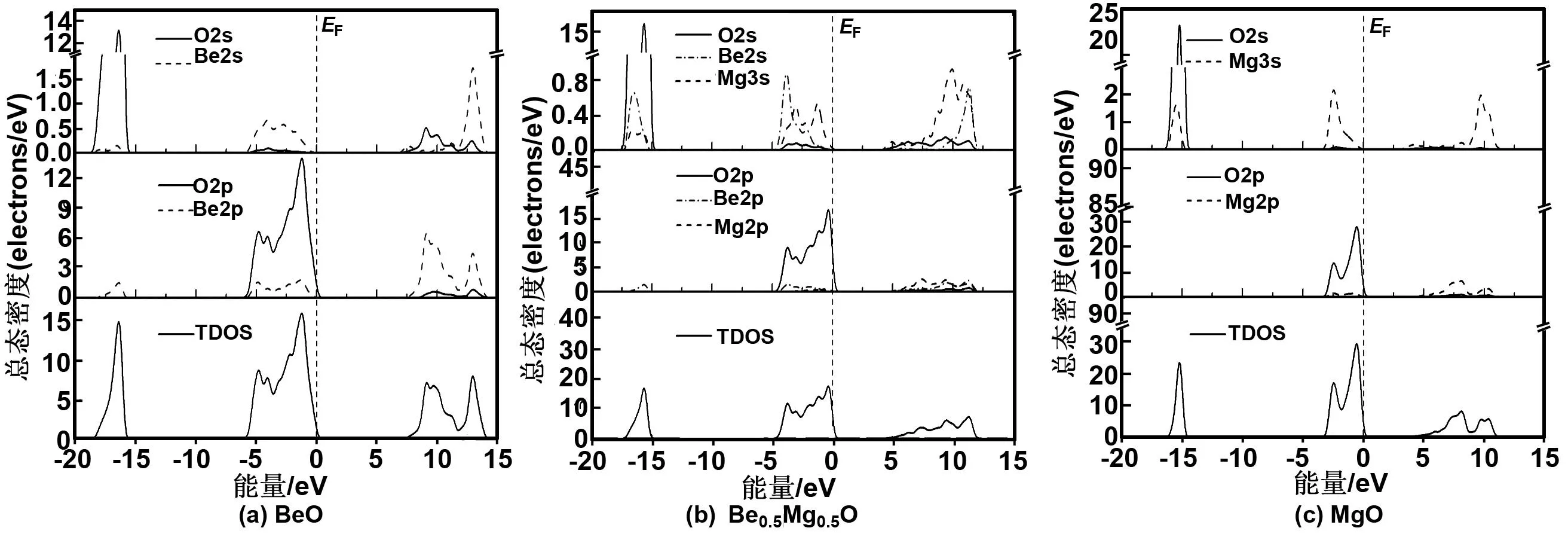
图2 纤锌矿BeO、Be0.5Mg0.5O和MgO的分波态密度和总态密度
Fig 2 Partial DOS and total DOS of w-BeO, w-Be0.5Mg0.5O and w-MgO, the Fermi level (EF) indicated by a vertical dashed line is set to be zero
2.2纤锌矿Be1-xMgxO合金的Milliken电荷分布
为了进一步理解Mg掺入对Be1-xMgxO合金中的各原子轨道电子分布和成键行为的影响,计算了纤锌矿BeO、Be0.5Mg0.5O和MgO材料中Be、Mg和O的Milliken电荷转移情况,见表2所示。
表2纤锌矿Be1-xMgxO(x=0,0.5,1)合金的Milliken电荷分布
Table 2 Calculated milliken charge population of w-Be1-xMgxO(x=0, 0.5, 1) alloys

原子s电子p电子总电荷净电荷BeOBe0.290.931.230.77O1.794.986.77-0.77Be0.5Mg0.5OBe0.461.161.620.38Mg0.405.896.281.72O*1.895.157.03-1.03O**1.885.197.07-1.07MgOMg0.466.256.711.29O1.965.337.29-1.29
注:O*代表该O原子周围的a方向为2个Be和1个Mg原子,在c方向上为1个Mg原子;O**代表该O原子周围的a方向为1个Be 和2个Mg原子,在c方向上为1个Be原子。
对于纤锌矿BeO,虽然计算所选取的Be电子组态为2s2,但是当Be与O原子在成键作用中,Be的s态和p态发生杂化,s态中有0.93个电子转移到p轨道上,还有0.77个电子转移到O的p态轨道上。由于O从Be中获得的净电荷数为0.77,小于其价电荷数2,所以Be与O之间的成键以共价键为主,还存在少量离子特性,这与Joshi等[26]通过康普顿光谱分析BeO成键得到实验结果相符。
对比BeO的电荷分布,Be0.5Mg0.5O合金的Be和O的s、p态电荷数都发生明显变化,这是因为Mg比Be能提供更多的轨道电子,致使Mg—O之间的净电荷转移数达到1.72,此时Be转移给O的净电荷数仅为0.38,比BeO时减少一半,但O原子获得的净电荷总数超过1,比BeO材料中O得到的电荷数多,更接近O的价电荷数,这表明Mg掺入BeO材料形成Be0.5Mg0.5O合金,合金中的成键整体特性由共价性逐步转向离子性,与之对应的是Be0.5Mg0.5O合金成键结合能和晶胞总能量逐步降低(见图3所示),这与图1分析得知当Mg掺杂组分增大,Be1-xMgxO合金能隙逐渐变窄的趋势相一致。 说明的是,表2中Be0.5Mg0.5O合金的每个O邻居都有2个Be和2个Mg,但纤锌矿结构的a方向键长小于c方向键长,造成O在键长差异下从Be和Mg获得的电荷数略有不同,但差别不大。对于纤锌矿MgO,Mg转移给O的电荷数达到1.29,进一步靠近O的价电荷数,表明Mg与O之间的成键偏向离子性,此时纤锌矿MgO的结合能和晶胞总能量进一步降低,能隙进一步变窄。

图3纤锌矿Be1-xMgxO(x=0,0.5,1)超原胞总能量和结合能
Fig 3 The total energy of super cells and binding energy (per pair) for w-Be1-xMgxO(x=0,0.5, 1) alloys
2.3纤锌矿Be1-xMgxO合金的相分离特性
2.3.1Be1-xMgxO合金的有序相
合金材料的有序和无序性是合金材料内部结构相的反映,研究合金的相分离问题就需要分析合金的有序无序相。对Mg掺杂BeO形成的Be1-xMgxO合金而言,Be1-xMgxO合金的有序相体现Mg掺杂取代Be位后所形成的合金,其结构内部的Be和Mg离子排序是按一定次序和方向进行分布(一般指短程有序分布),无序相是相对有序相而言。为了分析纤锌矿Be1-xMgxO合金的有序相,计算了有序结构的纤锌矿Be1-xMgxO(x=0,0.25,0.5,0.75,1)合金形成能。其形成能计算采用以下公式[27]
(1)
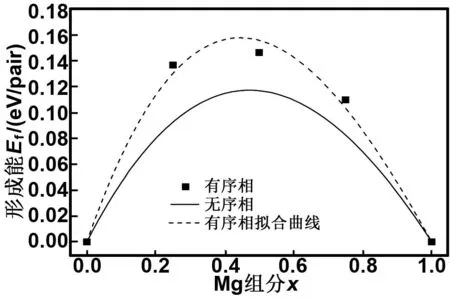
图4 有序相和无序相w-Be1-xMgxO合金形成能
Fig4Theformationenergyofw-Be1-xMgxOalloysfortheorderedanddisorderedphase
根据正规溶液理论[28]知,合金材料的形成能与组分关系可以表示为
(2)
式中,Ω为合金材料的相互作用能,跟不同材料体系相关,对于晶格振动效应不明显的合金材料,如GaAlAs材料,Ω为一个常数,而对于晶格振动效应明显的合金材料,Ω为一个与组分x相关的函数,一般表示为
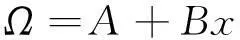
(3)
式中,A和B为两个参量。采用式(2)和(3)对图4形成能的计算值进行拟合,得到的拟合曲线(虚线)列于图4中,拟合得到的参量A和B值分别为0.765和-0.286 eV,这里B不为零,意味着纤锌矿Be1-xMgxO合金内部存在明显的晶格振动效应。由图4还得知,拟合曲线的最大形成能所对应Mg组分x值为0.44,与Mg组分的中心值0.5有一定偏离(晶格振动效应越明显,偏离就越大)。图5给出了不同Mg组分下Be1-xMgxO合金的Be—O和Mg—O平均键长值,由该图可知Be—O的平均键长在0.17 nm左右,而Mg—O的平均键长>0.195 nm,显然它们之间的键长值差距较大。文献[29]指出,纤锌矿合金材料的键长不同容易造成合金内部的晶格振动效应产生,因此纤锌矿Be1-xMgxO内部产生较强的晶格振动效应。
2.3.2Be1-xMgxO合金的无序相
对于纤锌矿Be1-xMgxO合金的无序相计算,采用Connolly和Williams等[30]的计算方法。 无序结构合金的形成能可采用以下的计算公式
(4)
其中
式中,Ef(x)为式(1)的Be1-xMgxO合金形成能。Pn(x)为统计上的权重因子,代表着Be1-xMgxO合金中第n个短程有序结构的概率。根据式(4)计算得到的无序结构Be1-xMgxO合金形成能曲线(实线)也绘于图4中。由图4得知,无序结构的Be1-xMgxO合金形成能低于有序结构的形成能,说明Mg的掺入更容易引起Be1-xMgxO合金的无序相产生,所以要想获得有序结构的Be1-xMgxO合金,就需要适当提高实验制备温度以使纤锌矿Be1-xMgxO合金有更多的掺杂激活能量。
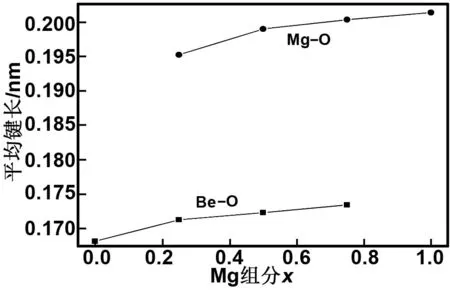
图5w-Be1-xMgxO合金的Be—O和Mg—O平均键长
Fig 5 Average bond length of Be—O and Mg—O in the w-Be1-xMgxO alloy
3结论
采用第一性原理的平面波赝势方法,研究了纤锌矿Be1-xMgxO合金的电子结构和相特性。结果表明,纤锌矿Be1-xMgxO合金的价带顶由O2p态决定,导带底由Mg3s态决定。当Mg掺杂组分增大,上价带宽度变窄、下价带和导带都朝价带顶方向移动,能隙逐渐变窄。由Milliken电荷分布知,Be与O之间主要以共价键结合,而Mg与O之间主要以离子键结合。当Mg的掺杂组分增多,Be1-xMgxO合金的离子性在增强,而结合能和总能量在降低。由于Be—O的平均键长比Mg—O平均键长明显小,造成纤锌矿Be1-xMgxO合金内部存在较强的晶格振动效应。无序结构的Be1-xMgxO合金形成能低于有序结构的形成能,表明Mg的掺入容易使纤锌矿Be1-xMgxO合金产生无序结构,因此在进行纤锌矿Be1-xMgxO合金的实验制备时,要适当增加实验温度。
参考文献:
[1]Bagnall D M, Chen Y F, Zhu Z, et al. Optically pumped lasing of ZnO at room temperature [J] Appl Phys Lett, 1997, 70(17):2230-2232.
[2]Park W I, Yi G C,Jang H M. Metalorganic vapor-phase epitaxial growth and photoluminescent properties of ZnMgO thin films[J]. Appl Phys Lett, 2001,79:2022-2024.
[3]Ohtomo A, Kawasaki M, Koida T, et al. MgxZn1-xO as a Ⅱ-Ⅵ wide gap semiconductor alloy[J].Appl Phys Lett, 1998, 72:2466-2468.
[4]Ryu Y R, Lee T S, Lubguban J A, et al. Wide-band gap oxide alloy:BeZnO [J]. Appl Phys Lett, 2006, 88:0521031.
[5]Kim W J, Leem T H, Han M S, et al. Crystalline properities of wide band gap BeZnO films [J]. J Appl Phys, 2006,99:0961041.
[6]Shi H L, Duan Y. Band-gap bowing and p-type doping of (Zn, Mg, Be)O wide-gap semiconductor alloys: a first-principles study[J]. Eur Phys J B, 2008,66:439-444.
[7]Zheng S W, Fan G H, Li S T, et al. Study on the energy band properties and phase stability of Be1-xMgxO alloy[J]. Acta Phys Sin, 2012,61(23):237101 1-9.
郑树文,范广涵,李述体,等.Be1-xMgxO合金的能带特性与相结构稳定性的研究[J].物理学报,2012,61(23):237101.
[8]Hou Qingyu, Zhao Chunwang. First principle studies of electronic structures and red shift effect heavily N-doped rutile TiO2[J]. Journal of Functional Materials, 2011, 42(5): 782-784.
候清玉,赵春旺.高掺杂N对金红石型TiO2电子结构和红移影响的理论研究[J].功能材料,2011,42(5):782-784.
[9]Zhu Liangdi, Zhang Jin, Zhu Zhongqi, et al.First-principles study on electronic structure and optical properties of anatase TiO2codoped with sulfur and manganese[J]. Journal of Functional Materials, 2013, 44(1): 22-27.
朱良迪,张瑾,朱忠其. S、Mn共掺杂锐钛矿相TiO2电子结构和光学性质的第一性原理研究[J].功能材料,2013,44(1):22-27.
[10]Jin X L, Lou S Y, Kong D G, et al. Investigation on the broadening of band gap of wurtzite ZnO by Mg-doping[J]. Acta Phys Sin, 2006,55(9): 4809-4815.
靳锡联,娄世云,孔德国,等. Mg掺杂ZnO所致的禁带宽度增大现象研究[J]. 物理学报, 2006, 55(9): 4809-4815.
[11]Shi L B, Li R B, Cheng S, et al. A study on electronic structure and optical properties of Zn1-xBexO [J]. Acta Phys Sin, 2009,58(9):6446-6451.
史力斌,李容兵,成爽,等. 关于Zn1-xBexO电子结构和光学性质的研究[J]. 物理学报, 2009,58(9):6446-6451.
[12]Kim W J, Leem T H, Han MS, et al. Crystalline properties of wide band gap BeZnO films[J]. J Appl Phys, 2006, 99:0961041.
[13]Zhu Y Z,Chen G D, Ye H, et al. Electronic structure and phase stability of MgO, ZnO, CdO, and related ternary alloys[J]. Phys Rev B, 2008,77:2452091.
[14]Hohenberg P, Kohn W. Inhomogeneous electron gas[J]. Phys Rev B, 1964, 136:864-871.
[15]Huang H C, Gilmer G H, Tomas D de la R. An atomistic simulator for thin film deposition in three dimensions[J].J Appl Phys, 1998,84:3636-3649.
[16]Perdew J P, Chevary J A,Vosko S H, et al. Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation[J]. Phys Rev B, 1992, 46:6671-6681.
[17]Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J].Phys Rev B, 1990,41:7892-7895.
[18]Monkhorst H J, Pack J D. Special points for brillouin-zone integrations-a reply[J]. Phys Rev B, 1977,16:1748-1749.
[19]Bouhalouane A,Fouad El H H, Hadi A. First-principles investigations of the ground-state and excited-state properties of BeO polymorphs[J].J Phys:Condens Matter, 2007,19: 436216-436227.
[20]Chang K J, Froyen S, Cohen M L. The electronic band structures for zincblende and wurtzite BeO[J].J Phys C, 1983, 16:3475-3480.
[21]Jephcoat A P, Hemley R J,Mao H K, et al. Raman spectroscopy and theoretical modeling of BeO at high pressure[J]. Phys Rev B, 1988,37(9):4727-4734.
[22]Schleife A, Fuchs F, Furthmüller J, et al. First-principles study of ground- and excited-state properties of MgO, ZnO and CdO polymorphs[J].Phys Rev B, 2006,73(24):245212 1-14.
[23]Wang Z J, Li S C, Wang L Y, et al. First-principles study of structrural and corrected band properties of wurtzite Zn1-xCdxO and Zn1-xMgxO systems[J].Chin Phys B, 2009,18(7):2992-2997.
[24]Chen K, Fan G H, Zhang Y, et al. First-principle calculation of nitrogen-doped p-type ZnO[J]. Acta Phys Chim Sin, 2008, 24(1): 61-66.
陈琨,范广涵,章勇,等. N掺杂p-型ZnO的第一性原理计算[J]. 物理化学学报,2008, 24(1):61-66.
[25]Li Q F, Tu Y, Tolner H, et al. Plasma discharge efficiency increase by using a small bandgap protective layer material- first-principles study for Mg1-xZnxO[J]. J Appl Phys, 2011, 109:093307 1-8.
[26]Joshi K B, Jain R, Pandya R K, et al. Compton profile study of bonding in BeO[J]. J Chem Phys, 1999,111(1):163-168.
[27]Sun H Q, Ding S F, Wang Y T, et al. Structural, Energetical and Electronic Properties of CdO and CdxZn1-xO compounds[J]. Acta Phys Chim Sin,2008,24(7):1233-1238.
孙慧卿,丁少峰,王雨田,等. CdO及CdxZn1-xO化合物的结构、能量和电子性能分析[J]. 物理化学学报,2008, 24(7): 1233-1238.
[28]Saito T, Arakawa Y. Atomic structure and phase stability of InxGa1-xN random alloys calculated using a valence-force- field method[J]. Phys Rev B, 1999, 60(3):1701-1706.
[29]Gan C K, Feng Y P, Srolovitz D J. First-principles calculation of the thermodynamics of InxGa1-xN alloys: effect of lattice vibrations[J].Phys Rev B, 2006, 73(23):235214 1-8.
[30]Connolly J W D, Williams A R. Density-functional theory applied to phase transformations in transition-metal alloys[J]. Phys Rev B, 1983,27(8):5169-5173.
Study on the electronic structures andphase properties of wurtzite Be1-xMgxO alloy
ZHENG Shuwen,ZHANG Tao,ZHANG Li, CHEN Zhenshi
(Guangdong Engineering Research Center of Optoelectronic Functional Materials and Devices, Institute of Opto-electronic Materials and Technology, South China Normal University, Guangzhou 510631,China)
Abstract:The electronic structures and phase properties of wurtzite(w) Be1-xMgxO alloy are investigated by the first-principles method based on the density functional theory. The calculated results show that the bandgap of w-Be1-xMgxO is determined by O2p and Mg3s states. The bandgap of Be1-xMgxO alloy becomes smaller and its ionic character is enhanced with the Mg content x of Be1-xMgxO is increased. There is a strong lattice vibration effect in the w-Be1-xMgxO structure, which chiefly originates from the large difference in the average bond length between Be—O and Mg—O. w-Be1-xMgxO alloy is likely to form disordered structure because the formation energy of its disordered phase is lower than its ordered phase, so it is necessary to prepared the w-Be1-xMgxO alloy with higher growth temperature.
Key words:first-principles; electronic structure; phase properties; Be1-xMgxO
DOI:10.3969/j.issn.1001-9731.2016.03.027
文献标识码:A
中图分类号:O469;O471
作者简介:郑树文(1977-),男,广东湛江人,副研究员,博士,主要从事宽能隙材料性能研究。
基金项目:国家自然科学基金资助项目(61176043);广东省科技计划资助项目(2015B090903078);华南师范大学青年教师培育基金资助项目(2012KJ018)
文章编号:1001-9731(2016)03-03146-05
收到初稿日期:2015-04-15 收到修改稿日期:2015-08-27 通讯作者:郑树文,E-mail:LED@scnu.edu.cn
