高尿酸血症治疗药物黄嘌呤氧化酶抑制剂的研究进展
2016-05-23陆海波鲁传华
陆海波,鲁传华
(安徽中医药大学药学院,安徽 合肥 230012)
高尿酸血症治疗药物黄嘌呤氧化酶抑制剂的研究进展
陆海波,鲁传华
(安徽中医药大学药学院,安徽 合肥230012)
摘要:高尿酸血症是由多种原因引起的代谢性疾病,黄嘌呤氧化酶(XO)是药物治疗高尿酸血症的重要靶点。随着黄嘌呤氧化酶晶体结构的解析、计算机辅助药物设计和高通量筛选等技术的运用,近年涌现了许多具有良好的XO抑制活性的化合物。中药也是我国治疗高尿酸血症的一种主要方式,对天然化合物进行结构修饰也是寻找新化合物的重要方法。该文基于高尿酸血症的产生机制,对具有良好XO抑制活性化合物进行综述。
关键词:高尿酸血症;痛风;尿酸;黄嘌呤氧化酶
随着现代人们生活方式的改变、药物的使用和人口的老龄化,高尿酸血症的发病率近年呈逐渐增长的趋势[1]。尿酸的过量产生和过少排泄会引发高尿酸血症,血尿酸的浓度≥417 μmol·L-1时被认为是高尿酸血症。
尿酸是嘌呤代谢的终产物,人体内嘌呤代谢如图1所示。血清中98%的尿酸以尿酸盐的形式存在,与细胞外液的钠离子形成溶解度较低的尿酸钠。长期高尿酸血症会导致尿酸钠晶体在关节、组织等处沉积,引发痛风等症状[2]。高尿酸血症也常伴发其他代谢性疾病,如高血压、心血管疾病和肾损伤等[3]。治疗高尿酸血症的关键在于降低血清中尿酸的含量。
尿酸主要在肝脏产生,通过肾脏排泄。经肾小管滤过的尿酸盐90%通过尿酸盐阴离子转运体介导重吸收,包括尿酸转运体1(URAT1)[4],因此抑制URAT1可增加尿酸的排泄。然而由于相关肝脏毒性的报道,此类药物如苯溴马隆的使用受到了限制[5],一种新的URAT1抑制剂RDEA-594最近通过了临床试验,被批准用于痛风的治疗。
黄嘌呤氧化酶(XO)催化次黄嘌呤和黄嘌呤分别氧化生成黄嘌呤和尿酸,通过抑制黄嘌呤氧化酶能减少尿酸的合成,进而降低血清中尿酸的浓度。XO的生物作用、机制和结构明确,靶向XO的抑制剂是降尿酸药物研究的重点。

图1 体内嘌呤代谢简易图
1XO的结构和催化机制
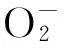
黄嘌呤氧化还原酶由一个同源二聚体组成,每个亚基有三个结构域:一个钼辅因子(Mo),两个铁硫中心(2Fe-2S)和一个黄素腺嘌呤二核苷酸(FAD)。氧化反应发生在钼中心附近,钼中心的结构如图2所示,通过两个S原子与蝶呤相连,以Mo=O中的O为顶点形成一个四面锥体的结构[8]。
次黄嘌呤和黄嘌呤作为底物与酶活性位点结合模式如图3,4所示,其中氨基酸残基Glu802和Arg880(牛乳XO的氨基酸顺序)可以与底物形成氢键,促进催化反应的进行[9]。
黄嘌呤氧化酶抑制剂(XOI)结合在Mo中心的催化活性位点,抑制黄嘌呤氧化酶,可阻断底物的羟化反应,减少尿酸的产生,从而降低血尿酸的浓度。
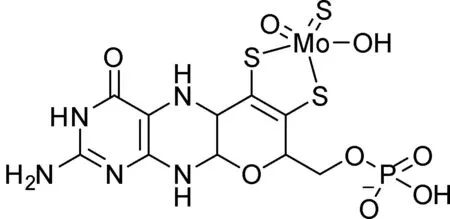
图2 钼蝶呤中心结构
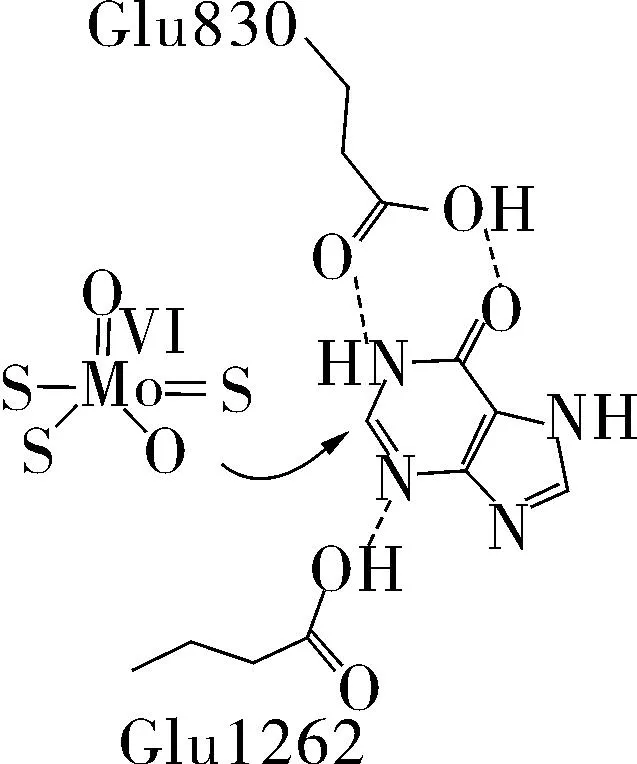
图3 推测的次黄嘌呤的结合模式

图4 推测的黄嘌呤的结合模式
2已上市和进入临床试验的黄嘌呤氧化酶抑制剂
2.1基于机制的黄嘌呤氧化酶抑制剂——别嘌醇别嘌醇是黄嘌呤氧化酶的抑制剂,该药物由Elion等[10]发明于20世纪60年代,是第一个上市的XOI,应用于阻断尿酸的产生以治疗高尿酸血症。
最初认为别嘌醇是竞争性的抑制剂,后来发现别嘌醇的羟化产物别黄嘌呤是真正的强效抑制剂。研究人员通过分析别黄嘌呤和酶的晶体复合物,发现Mo原子和N-2之间形成了共价结合,此外别黄嘌呤4位和6位的羰基分别与Glu802和Arg880形成氢键,N-1也可与Glu1261形成氢键(图5),使得化合物和酶的结合非常牢固[11]。Glu802、Arg880和Glu1261是催化底物所必需的氨基酸残基,别黄嘌呤通过在这个位点的作用,有效地抑制了酶的活性,阻断了底物的羟化反应。

图5 黄嘌呤氧化酶在活性位点和别黄嘌呤相互作用示意图
别嘌醇自上市以来广泛用于高尿酸血症和痛风的治疗,然而应用别嘌醇治疗高尿酸血症会导致至少2%的患者出现别嘌醇过敏综合征(AHS),包括中毒性表皮坏死松解症、肝炎、间质性肾炎等症状[12],别嘌醇可能引发的严重副作用限制了该药物的使用。
2.2基于结构的黄嘌呤氧化酶抑制剂
2.2.1非布索坦非布索坦(Febuxostat,TMX-67,TEI-6720)是一种强效的黄嘌呤氧化酶抑制剂,2008年率先在欧洲上市,2009年通过FDA的批准,用于治疗高尿酸血症和痛风[13]。
非布索坦具有复合的酶抑制作用,抑制常数Ki值为0.9 nmol·L-1。通过非布索坦和黄嘌呤脱氢酶复合物的晶体结构发现化合物在外部溶剂到Mo蝶呤辅酶的通道位置与酶链接[14]。非布索坦的羧基、氰基分别和Arg880、Glu802和Asn768等氨基酸残基形成氢键(图6)。噻唑环位于Phe914和Phe1009之间,产生π-π相互作用。此外,异丁基侧链被Leu648,Phe649和Val1011和Leu1013围绕,产生疏水作用。这些位于细长接入通道的相互作用,使非布索坦和氨基酸残基形成非常紧密的连接。

图6 非布索坦与黄嘌呤氧化酶的相互作用
临床试验显示[15],高尿酸血症患者在以80 mg·d-1的剂量接受的非布索坦治疗3个月后,有53%的患者血尿酸浓度降至6.0 mg·dL-1以下,明显高于接受300 mg·d-1剂量的别嘌醇治疗组(21%)。使用非布索坦治疗的患者副作用发生率低于2%,主要是肝酶升高、皮疹、关节疼痛等[16]。非布索坦在降尿酸的疗效和副作用方面明显优于别嘌醇,现在临床被广泛用于高尿酸血症和痛风的治疗。
2.2.2Y-700别嘌醇和别黄嘌呤是磷酸转移酶的底物,形成N-1核糖衍生物,别黄嘌呤还会被尿苷磷酸化酶转换成N-7核糖衍生物,这是别嘌醇长期给药过程中可能的毒性物质[17]。Ishibuchi等[18]通过如下方案对别嘌醇进行结构改造(图7):(1)在N-1位引入苯环,阻断在N-1位被转化成非天然核苷酸衍生物的可能;(2)用羧基替代4位的羟基嘧啶部分,希望可以增加与催化中心Mo的相互作用。
通过对系列化合物进行的活性研究,发现化合物1和Y-700(图8)具有最佳的体外抑制活性,半抑制浓度IC50分别为7.1 nmol·L-1和5.8 nmol·L-1。在氧嗪酸钾诱导的高尿酸血症动物模型降尿酸实验中,Y-700显示了优于别嘌醇的强效且持久的降尿酸作用。
Y-700对黄嘌呤氧化酶呈现混合型的抑制作用,Ki=0.6 nmol·L-1,Ki'=3.2 nmol·L-1。药效动力学实验发现,Y-700具有剂量依赖的降尿酸作用,并且在口服10 h之后依然有效, Y-700是一类强效且作用持久的黄嘌呤氧化酶抑制剂[19]。
2.3混合机制的抑制剂—托匹司他 Sato等[20]发现三唑类化合物2、3具有良好的IC50值和体内降尿酸活性,然而其药物代谢动力学性质不佳,如最大血药浓度值(Cmax),分别只有16.7 、712 μg · L-1。

图7 不含嘌呤核的黄嘌呤氧化酶抑制剂设计路线
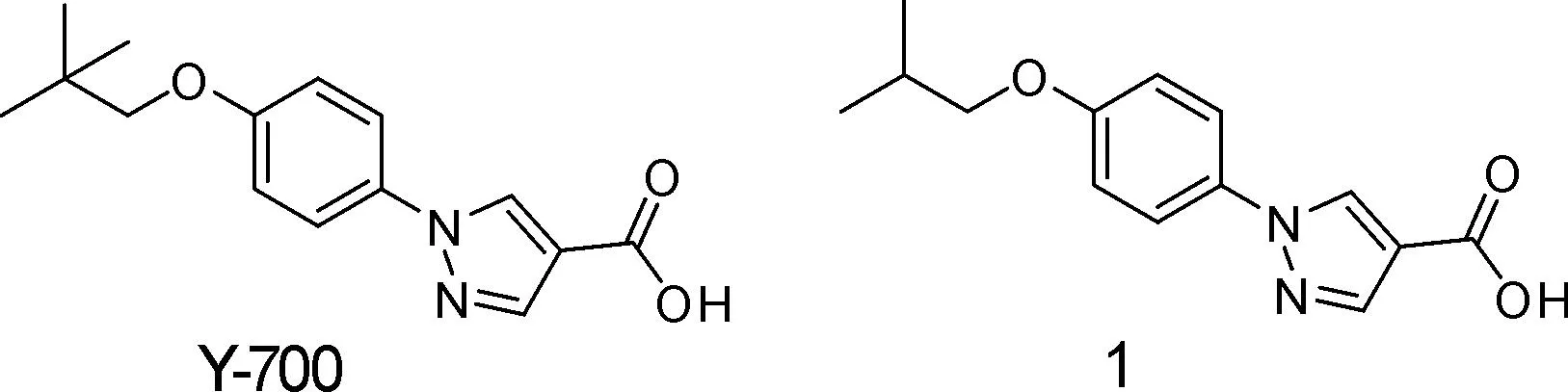
图8 化合物1和Y-700的结构式
研究人员注意到化合物4具有较高的Cmax[21],同时在吡啶基的2位引入甲基,可以有效增加体内的活性。通过保留2-甲基-4-吡啶-三唑部分,在另一个吡啶的2位引入氰基得到的化合物5具有更好的降尿酸作用和更高的Cmax值,达到3.97 mg · L-1。再对化合物5的甲基部分进行修饰,发现没有取代基的化合物6(FYX-051)具有最佳的降尿酸作用。同时化合物6的半衰期为19.7 h,具有良好的药物代谢动力学性质(图9)。
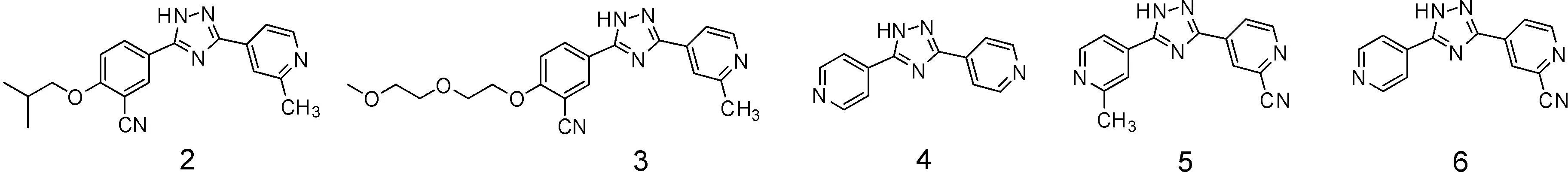
图9 化合物2、3、4、5、6的结构式
FYX-051最初在酶的活性位点形成Mo-O-C的共价连接,同时与周围的氨基酸残基产生相互作用,是一种具有混合机制的XOI[9]。Matsumoto等[22]进一步发现,FYX-051和酶作用后会氧化成三羟基化的产物,通过氰基和Mo原子形成Mo-N-C的连接,产生持久的酶抑制作用。
3其他在研的具有新颖结构和良好酶抑制活性的化合物
3.1噻唑类Song等[23]通过电子等排原理,将非布索坦的苯环替换成吲哚,得到化合物7的IC50值为110 nmol·L-1,选作苗头化合物。在吲哚的3位引入吸电子基团后,IC50值显著降低,其中化合物8的IC50值达到5.5 nmol·L-1,同时具有较好的代谢稳定性,半衰期(T1/2)为105 min,它的结构类似物9的IC50值为3.5 nmol·L-1,具有较低的细胞毒性,和良好的药物代谢动力学性质。化合物7~9结构见图10。
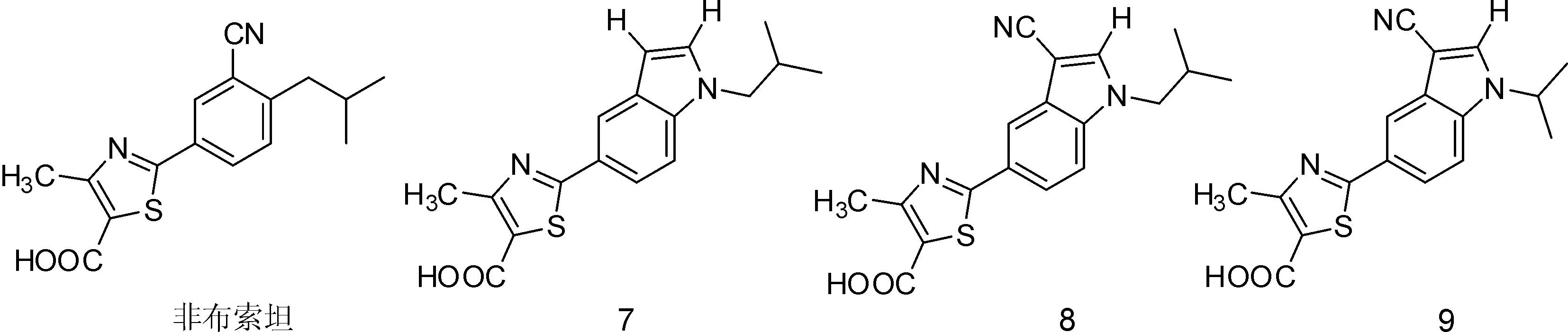
图10化合物7、8、9的结构式
对化合物9进行进一步的结构修饰,发现多数化合物保持了良好的XO的抑制活性,但体内的降尿酸作用对比化合物9较弱。化合物9以10 mg·kg-1对高尿酸模型大鼠口服给药,1 h后尿酸的抑制率为60%,Cmax=3.98 mg·L-1, T1/2=7.95 h,生物利用度为84.31%,化合物9有望进一步进入临床研究。
3.2咪唑类 Chen等[24]设计合成了一系列1-羟基/甲氧基-4-甲基-2-苯基-1H-咪唑-5-羧酸类化合物。1-羟基取代的化合物显示了较强的酶抑制活性,1-甲氧基取代的化合物多数没有表现出活性(如化合物13),表明1位羟基和化合物的效能有重要的关联。1-羟基系列化合物中,苯环4’位分别用正丁氧基、仲丁氧基和异丁基取代的化合物10,11和12(图11),IC50值分别为0.003 μmol·L-1、0.003 μmol·L-1和0.006 μmol·L-1,优于非布索坦的0.01 μmol·L-1。当4’位用其他烷氧基取代活性均有所下降(化合物13)。
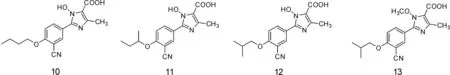
图11 化合物10、11、12、13的结构式
化合物10、11和12具有强效的XO抑制活性,可作为治疗痛风、高尿酸血症及其他关联黄嘌呤氧化酶疾病的候选化合物。
4天然化合物的衍生物
对天然化合物进行结构修饰,用来提高活性和改善药物代谢性质,是一种寻找新型化合物的重要方法。
Singh等[30]将具有XO抑制活性的萘吡喃骨架和黄酮类化合物骨架拼合,设计和合成了一系列萘黄酮类化合物,其中化合物14(图12)具有最好的活性,IC50值为0.62 μmol·L-1。Niu等[31]通过对异甘草素的衍生化,得到化合物15具有较好的体内降尿酸作用,并且没有明显的毒性反应。

图12 化合物14、异甘草素和化合物15的结构式
芹菜素具有良好的XO抑制活性,由于较低的生物利用度,临床使用效果不佳。羧基广泛运用于临床使用的药物中,Su等[32]通过引入羧基(图13),期望改善化合物的活性和代谢动力学性质。酶抑制实验的结果表明,在4’位引入羧烷基,化合物的XO抑制活性明显提高,化合物16的IC50值达到0.098 μmol·L-1,比芹菜素的活性提高30倍。然而在5、7位的羧烷基衍生物没有XO的抑制活性。实验显示化合物16具有降尿酸作用,但仍弱于别嘌醇。

图13 芹菜素衍生物的设计策略
5总结
近年高尿酸血症和痛风的发病率逐渐上升,给人们的生活带来了许多的痛苦和不便。同时,高尿酸血症与心血管疾病、糖尿病和肾脏疾病显著相关,降尿酸药物受到了越来越多的关注。随着高尿酸血症产生的原因逐步明确,以及黄嘌呤氧化酶结构被逐步解析,研究人员通过生物电子等排和计算机辅助药物设计等方法,设计合成了许多具有良好XO抑制活性的化合物。研究人员从临床用于治疗痛风的中药和天然植物中,发现了一些具有XO抑制作用的天然化合物,虽然活性相对不高,但普遍具有较好的安全性,也可作为先导化合物,通过进一步的结构修饰,以提高选择性和活性,用于高尿酸血症和痛风的治疗。
参考文献:
[1]Richette P,Bardin T.Gout [J].Lancet,2010,375(9711):318-328.
[2]Robinson PC,Simon H.Gout:joints and beyond,epidemiology,clinical features,treatment and co-morbidities [J].Maturitas,2014,78(4):245-251.
[3]邹筱芳,巫冠中.尿酸肾损伤的分子机制研究进展 [J].安徽医药,2015,19(1):5-9.
[4]Bobulescu IA,Moe OW.Renal transport of uric acid: evolving concepts and uncertainties [J].Adv Chronic Kidney Dis,2012,19(6):358-371.
[5]Robert T.Update on gout:new therapeutic strategies and options [J].Nat Rev Rheumatol,2010,6(1):30-38.
[6]Parks DA,Granger DN.Xanthine oxidase: biochemistry,distribution and physiology [J].Acta Physiol Scand Suppl,1986,548(1):87-99.
[7]Okamoto K,Kusano T,Nishino T.Chemical Nature and Reaction Mechanisms of the Molybdenum Cofactor of Xanthine Oxidoreductase [J].Curr Pharma Design,2013,19(14):2606-2614.
[8]Okamoto K,Matsumoto K,Hille R,et al.The crystal structure of xanthine oxidoreductase during catalysis: implications for reaction mechanism and enzyme inhibition [J].Proc Natl Acad Sci USA,2004,101(21):7931-7936.
[9]Yuichiro Y,Tomohiro M,Kimiyoshi I,et al.Human xanthine oxidase changes its substrate specificity to aldehyde oxidase type upon mutation of amino acid residues in the active site: roles of active site residues in binding and activation of purine substrate [J].J Biochem,2007,141(4): 513-524.
[10] Elion GB.The purine path to chemotherapy [J].Science,1989,244(4900): 509-529.
[11] Okamoto K,Eger BT,Nishino T,et al.Mechanism of Inhibition of Xanthine Oxidoreductase by Allopurinol: Crystal Structure of Reduced Bovine Milk Xanthine Oxidoreductase Bound with Oxipurinol [J].Nucleosides,2008,27(6):888-893.
[12] Hande KR,Noone RM,Stone WJ.Severe allopurinol toxicity: Description and guidelines for prevention in patients with renal insufficiency [J].Am J Med,1984,76(1):47-56.
[13] Burns CM,Wortmann RL.Gout therapeutics: new drugs for an old disease [J].Lancet,2011,377(9760):165-177.
[14] Okamoto K,Eger BT,Nishino T,et al.An Extremely Potent Inhibitor of Xanthine Oxidoreductase [J].J Biol Chem,2003,278(3):1848-1855.
[15] Ernst M,Fravel M.Febuxostat:a selective xanthine-oxidase/xanthine-dehydrogenase inhibitor for the management of hyperuricemia in adults with gout [J].Clinical Therapeutics,2009,31(11): 2503-2518.
[16] Felser A,Lindinger PW,Schnell D,et al.Hepatocellular toxicity of benzbromarone: Effects on mitochondrial function and structure [J].Toxicology,2014,324:136-146.
[17] Krenitsky TA,Elion GB,Strelitz RA,et al.Ribonucleosides of allopurinol and oxoallopurinol Isolation from human urine,enzymatic synthesis,and characterization [J].J Biol Chem,1967,242(11): 2675-2682.
[18] Ishibuchi S,Morimoto H,Oe T,et al.Synthesis and structure-activity relationships of 1-phenylpyrazoles as xanthine oxidase inhibitors [J].Bioorg Med Chem Lett,2001,11(7):879-882.
[19] Fukunari A,Okamoto K,Nishino T,et al.Y-700 [1-[3-Cyano-4-(2,2-dimethylpropoxy) phenyl]-1H-pyrazole-4-carboxylic acid]: a potent xanthine oxidoreductase inhibitor with hepatic excretion[J].J Pharmacol Exp Ther,2004,311(2): 519-528.
[20] Sato T,Ashizawa N,Iwanaga T,et al.Design,synthesis,and pharmacological and pharmacokinetic evaluation of 3-phenyl-5-pyridyl-1,2,4-triazole derivatives as xanthine oxidoreductase inhibitors[J].Bioorg Med Chem Lett,2009,19(1):184-187.
[21] Sato T,Ashizawa N,Matsumoto K,et al.Discovery of 3-(3-cyano-4-pyridyl) -5-(4-pyridyl) -1,2,4-triazole,FYX-051-a xanthine oxidoreductase inhibitor for the treatment of hyperuricemia [J].Bioorg Med Chem Lett,2009,19(21):6225-6229.
[22] Matsumoto K,Okamoto K,Ashizawa N,et al.FYX-051: a novel and potent hybrid-type inhibitor of xanthine oxidoreductase [J].J Pharmacol Exp Ther,2011,336(1): 95-103.
[23] Song JU,Choi SP,Kim TH,et al.Design and synthesis of novel 2-(indol-5-yl)thiazole derivatives as xanthine oxidase inhibitors [J].Bioorg Med Chem Lett,2015,25(6):1254-1258.
[24] Chen S,Zhang TJ,Wang J,et al.Synthesis and evaluation of 1- hydroxy/methoxy- 4- methyl- 2- phenyl- 1H- imidazole-5-carboxylic acid derivatives as non-purine xanthine oxidase inhibitors [J].Eur J Med Chem,2015,103:343-353.
[25] Nguyen MT,Awale S,Tezuka Y,et al.Xanthine oxidase inhibitory activity of Vietnamese medicinal plants [J].Biol Pharm Bull,2004,27(9):1414-1421.
[26] Lin CM,Chen CS,Chen CT,et al.Molecular modeling of flavonoids that inhibits xanthine oxidase [J].Biochem Bioph Res Co,2002,294(1):167-172.
[27] Cao H,Pauff JM,Hille R.X-ray crystal structure of a xanthine oxidase complex with the flavonoid inhibitor quercetin [J].J Nat Prod,2014,77(7): 1693-1699.
[28] Liu HX,He MT,Tan HB,et al.Xanthine oxidase inhibitors isolated from Piper nudibaccatum [J].Phytochem Lett,2015,12:133-137.
[29] Lin HC,Tsai SH,Chen CS,et al.Structure-activity relationship of coumarin derivatives on xanthine oxidase-inhibiting and free radical-scavenging activities [J].Biochem Pharmacol,2008,75(6): 1416-1425.
[30] Singh H,Sharma S,Ojha R,et al.Synthesis and evaluation of naphthoflavones as a new class of non purine xanthine oxidase inhibitors [J].Bioorg Med Chem Lett,2014,24(17):4192-4197.
[31] Niu YF,Zhu HJ,Liu J,et al.3,5,2',4'-Tetrahydroxychalcone,a new non-purine xanthine oxidase inhibitor [J].Chem-Biol Interact,2011,189(3):161-166.
[32] Su ZR,Fan SY,Shi WG,et al.Discovery of xanthine oxidase inhibitors and/or α-glucosidase inhibitors by carboxyalkyl derivatization based on the flavonoid of apigenin [J].Bioorg Med Chem Lett,2015,25(14):2778-2781.
Research process of xanthine oxidase inhibitors as hyperuricemia drug
LU Hai-bo,LU Chuan-hua
(SchoolofPharmacy,AnhuiUniversityofChineseMedicine,Hefei,Anhui230012,China)
Abstract:Hyperuricemia is a metabolic disease resulting from multiple factors,and xanthine oxidase (XO) is an important target for hyperuricemia therapy.Due to the determination of crystal structure of the enzyme and application of CADD and HTS,there emerge a large number of xanthine oxidase inhibitors which have good activity for lowering uric acid in recent years.Using traditional Chinese medicine to treat hyperuricemia is prevalent in China. It is an important method to modificate natural product to find new compounds.In this article,we review the compounds which have excellent inhibitory activity of xanthine oxidase based on the pathological mechanism.
Key words:hyperuricemia;gout;uric acid;xanthine oxidase
(收稿日期:2015-11-19,修回日期:2016-01-15)
doi:10.3969/j.issn.1009-6469.2016.04.004
作者简介:陆海波,男,硕士研究生通信作者:鲁传华,男,教授,硕士生导师,研究方向:药用新材料及其药物制剂新技术,E-mail:lchchem@126.com
