耕种模式对三江平原黑土细菌多样性的影响
2016-05-14汪城墙黄峤璟吕刚胡文豪朱瑞敏丁延芹杜秉海
汪城墙 黄峤璟 吕刚 胡文豪 朱瑞敏 丁延芹 杜秉海

摘要:本试验使用Illumina MiSeq测序平台,选取我国东北三江平原地区的大田黑土、菜园黑土和森林黑土3种样本,对不同耕种黑土表层细菌群落结构进行分析。结果显示:大田土、菜园土和森林土的微生物群落存在明显差异。大田土的群落丰富度明显低于菜园土和森林土,后两者相似度较高。大田土的OTU数比菜园土和森林土分别少13.3%和7.6%。菜园土的微生物群落组成种类最为丰富,森林土中硝化螺旋菌门含量最高,大田土微生物群落含有的外源性物质降解相关基因相对丰度较高。可见,三江平原大田黑土的退化和土壤微生物群落的改变有关,调节土壤微生物群落结构将有助于东北大田黑土的修复。
关键词:细菌群落多样性;高通量测序;黑土地;耕种模式
中图分类号:S154.36文献标识号:A文章编号:1001-4942(2016)07-0076-06
我国东北地区的黑土为世界上四大黑土地之一,是我国主要土壤类型之一,其土质肥沃、质地疏松,是重要的商品粮生产基地[1,2],总面积达1.0×107 hm2[3],由黑龙江、松花江和乌苏里江冲积而成的三江平原是其中的重要组成部分。但由于我国长期实行的农业耕作方式产生的一些弊端,尤其是长期单独施用化肥,致使黑土肥力显著下降,出现土壤板结、酸化、可耕性变差等土壤退化问题[4,5],亟待解决。
土壤含有大量的微生物,包括细菌、放线菌、真菌、原生动物和显微藻类等,其中细菌含量最高[6],在黑土土壤中约占微生物总数的80%以上[7]。土壤微生物群落是土壤生态系统的重要组成部分,在土壤有机物质转化和养分循环等多种生态过程中起着积极作用[8]。土壤微生物群落对生存环境的变化十分敏感,可以随着环境变化做出响应[9],是衡量土壤生物学活性以及生产力的一项重要指标[3]。土壤微生物群落多样性即为土壤微生物活性指标中最重要的因素[10],在一定程度上反映土壤性能。有研究表明,长期不同的人为施肥制度对土壤的微生物数量和群落结构影响显著[11],如:长期施入氮肥会降低土壤微生物活性;而植物残体和粪肥等有机肥料的施入可维持土壤肥力及微生物系统稳定,进而可改善土壤微生态环境或提高土壤微生物群落功能多样性[4,12,13]。对黑土土壤微生物群落功能多样性进行研究,可分析出微生物群落的功能,找出土壤肥力和土壤微生物之间的关系,对黑土土壤肥力及健康评价具有重要意义[2],也可对退化黑土土壤进行有效的微生物修复提供重要参考。近年来,对土壤微生物的群落结构与组成的研究备受关注。
本试验研究了长期施用化肥并种植经济作物的大田表层黑土、长期施用农家肥并种植蔬菜的菜园表层黑土以及森林表层黑土的细菌群落多样性,分析长期不同耕种方式及人为干扰对三江平原黑土地表层细菌群落多样性的影响,为三江平原大田黑土的微生物修复策略找到合适的微生物菌群、实现土地可持续利用提供科学依据。
1材料与方法
1.1取样
取样地点分布在黑龙江省鹤岗市萝北县军川农场(隶属北大荒集团),地处三江平原,介于东经131°02′~131°30′、北纬47°20′~47°40′之间。选取样本分为三类:森林土、菜园土以及大田土,分别编号为Ne1、Ne3和Ne4。森林土为东北寒温带原始森林地表层以下10~15 cm的黑土,菜园土为20世纪70年代开垦以来长期混种多种蔬菜和不定期施用不同种类农家肥的地表层以下10~15 cm的黑土,大田土为开垦以来长期种植大豆和玉米两种作物并施用化肥的地表层以下10~15 cm的黑土。取样时间为2015年8月1日。土壤取出后立即于-20℃冻存,一个月内完成分析工作。
1.2样品处理
每种样本选择两个生物学重复,用美国OMEGA公司的E.Z.N.A. Soil DNA Kit提取土壤总DNA,检测合格后,采用Illumina MiSeq测序平台,委托上海派森诺生物科技股份有限公司进行土壤细菌多样性检测。
2结果与分析
2.1样本测序数量的可信度
本试验中测序分析选择的测序数量为40 000条左右,从图1稀释曲线可以看出,所选测序斜率明显变小,说明测定的各样本能够反映真实物种的丰富度,数据分析和处理合理、可信度高。
2.2Alpha多样性分析
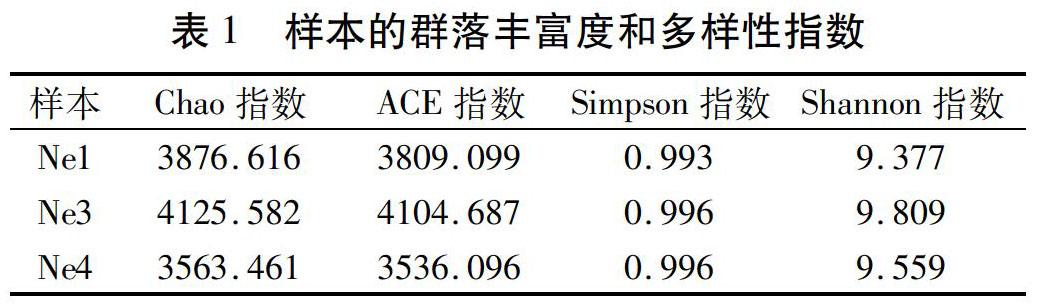
一个特定区域或生态系统内的微生物多样性常用Alpha 多样性来表示,可反映出丰富度和均匀度的综合指标。群落丰富度的指数Chao指数和ACE指数均和样本群落丰富度呈正比。群落多样性的指数Shannon指数和Simpson指数,分别和群落多样性呈正比和反比。三种样本在97%相似水平上的Alpha 多样性分析结果见表1。样本Ne3的Chao 指数和 ACE 指数最大,说明样本中含有的OTU数多,即群落丰富度最高;其次是Ne1,最小的是Ne4。不同样本的Simpson指数差别不大,Shannon指数具有Ne3、Ne4、Ne1依次减小的趋势,群落多样性逐渐降低。
2.3Beta多样性分析
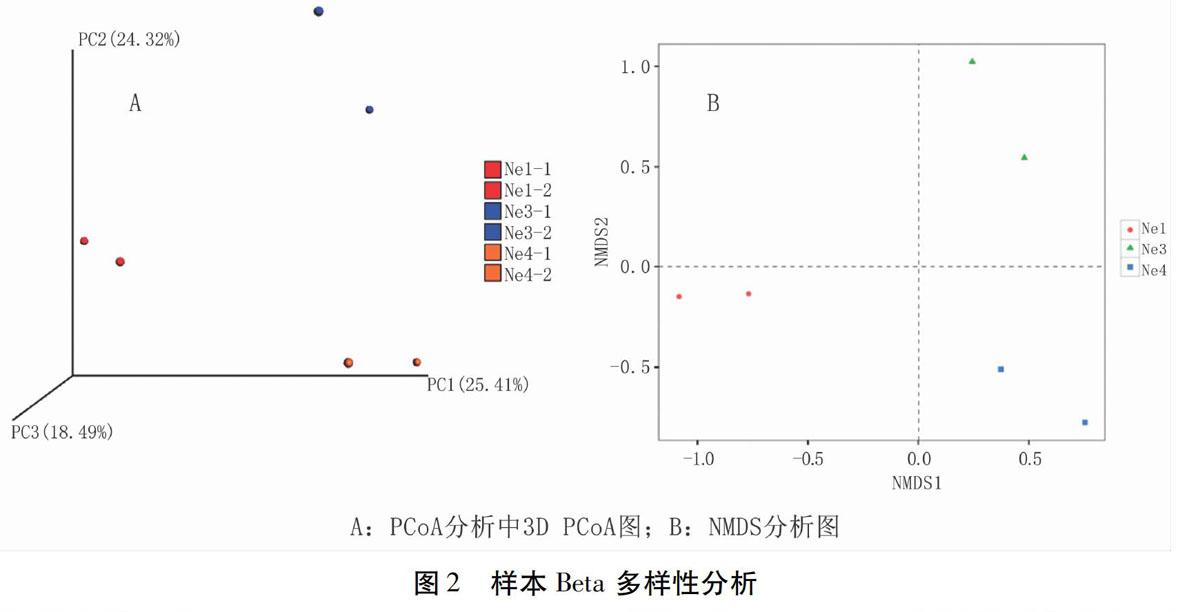
Beta 多样性是不同生态系统之间的多样性比较,可反映样本间是否具有显著的微生物群落差异。本试验使用Qiime软件对三组样本之间物种的进化以及丰度信息进行 Unifrac分析[14],得到样本间差异的距离矩阵,然后对样本之间的距离矩阵信息进行 PCoA 分析和 NMDS 分析,对PCoA分析的结果绘制出3D 图(图2A),对 NMDS 分析的结果利用 R 软件绘制出NMDS 图(图2B)。图中的每个点代表一个样本,颜色相同的点属于同一种分组,三样本组的两个生物学重复之间的距离较近,三种样本间的距离相对较远,说明三种样本间的微生物群落存在差异。
2.4样本群落组成分析
根据测序得到的 OTU结果,可以得到各种样本在各分类水平上,即门、纲、目、科、属上的物种组成比例情况,反映样本在不同分类学水平上的群落组成结构。Ne1、Ne3和Ne4三种样本中检测到的细菌门数分别为38、40、39;细菌纲数分别为115、119、109;细菌目数分别为166、168、161;细菌科数分别为200、213、202;细菌属数分别为241、270、257;细菌种数分别为78、87、81。在各分类水平上,Ne3都表现出最多的数量;在纲、目水平上,Ne1比Ne4表现出一定数量优势;在科、属和种水平上,Ne4比Ne1表现出数量优势。
由图3可以看出,在Ne1、Ne3和Ne4三组样本中,变形菌门(Proteobacteria)、放线菌门(Actinobacteria)和拟杆菌门(Bacteroidetes)含量最多,分别占到60.5%、63.75%、66.39%。疣微菌门(Verrucomicrobia)在Ne1、Ne3和Ne4中差异显著,含量分别为10.02%、3.33%和3.08%。酸杆菌门(Acidobacteria)含量差别也较大,在Ne1、Ne3和Ne4中的含量分别为6.33%、7.68%和4.92%。绿弯菌门(Chloroflexi)和浮霉菌门(Planctomycetes)在三个样本Ne1、Ne3和Ne4中含量具有逐渐增多趋势。Ne1中的硝化螺旋菌门(Nitrospirae)含量为0.75%,明显高于Ne3和Ne4中的0.35%和0.17%;Ne1中的热袍菌门(Thermotogae)也明显高于Ne3和Ne4;而厚壁菌门(Firmicutes)的含量只为Ne3和Ne4的约一半。Ne3中绿菌门(Chlorobi)含量明显低于Ne1和Ne4,而衣原体门(Chlamydiae)和梭杆菌门(Fusobacteria)的含量明显高于Ne1和Ne4,芽单胞菌门(Gemmatimonadetes)的含量也略高于Ne1和Ne4。Ne4中螺旋体门(Spirochaetes)较高于Ne1和Ne3。另外,在Ne1中未检测到纤维杆菌门(Fibrobacteres)和互养菌门(Synergistetes),在Ne4中未检测到脱铁杆菌门(Deferribacteres),总体上来看,菜园土Ne3中的细菌门水平上种类最为丰富。
2.5样本间的OTU 聚类分析
本试验利用R软件在97%的相似水平上对样本Ne1、Ne3和Ne4序列进行 OTU 的聚类分析,生成样本间OTU 的维恩图,分析菌种数目[10],结果见图4。样本Ne1、Ne3和Ne4中的OTU数分别为4 498、4 791和4 156。Ne4的OTU数比Ne1和Ne3分别减少7.6%和13.3%,Ne3的比Ne1高6.5%。样本Ne1、Ne3和Ne4共有的OTU数为2 351。样本Ne1、Ne3和Ne4特有的OTU数分别为754、872和568。Ne4和Ne3共有OTU数为3 057,和Ne1共有2 882。Ne1和Ne3共有OTU数为3 213,明显高于两样本和Ne4的共有数量。
应用软件R将三组样本属水平上的分类信息进行聚类,绘制 heatmap 热图,见图5。热图 heatmap用颜色变化直观地将数据值大小表示出来,将高丰度和低丰度的物种分块聚集,反映三样品在属水平上群落组成的相似性和差异性。结果显示,相比于人类耕种的样本Ne3和Ne4,天然样本Ne1的甲基杆菌属(Methylobacterium)、特吕珀菌属(Truepera)、苍白杆菌属(Ochrobactrum)、拉氏杆菌属(Rathayibacter)、Azohydromonas和黄杆菌属(Flavobacterium)的物种丰度较高。相比于大田样本Ne4,样本Ne1和Ne3的新鞘脂菌属(Novosphingobium)、气微菌属(Aeromicrobium)、红游动菌属(Rhodoplanes)、特吕珀菌属(Truepera)、苍白杆菌属(Ochrobactrum)、拉氏杆菌属(Rathayibacter)、Azohydromonas和黄杆菌属(Flavobacterium)丰度较高。相比于Ne1和Ne4,菜园土样本Ne3的Kaistobacter、出芽菌属(Gemmata)、农杆菌属(Agrobacterium)、丰佑菌属(Opitutus)、气微菌属(Aeromicrobium)、红游动菌属(Rhodoplanes)丰度较高。不同样本之间细菌属的类别存在较大差异。大田土Ne4与菜园土Ne3、森林土Ne1的相似度较低,而菜园土Ne3和森林土Ne1的相似度较高。
2.6群落功能基因预测
基于宏基因组的物种丰度信息以及物种的基因组数据库信息,预测三样品群落的功能基因组成。根据OTU水平的丰度信息,利用软件PICRUSt[15]进行功能基因的预测和相关统计分析,更加深入地了解样本的微生物群落并可比较样本在功能基因方面的差异,结果见图6。
转录相关基因、折叠分拣以及讲解相关基因、质代谢相关基因、多糖生物合成和代谢相关基因有由样本Ne1、Ne3和Ne4相对丰度逐渐减少的趋势。外源性物质降解和代谢相关基因、氨基酸代谢相关基因有由样本Ne1、Ne3和Ne4相对丰度逐渐增加的趋势。Ne3样本中,细胞运动性相关基因、复制和修复相关基因相对丰度较高,膜运输基因较少。
3讨论与结论
我国大田耕作中,大量施用化肥以及长期耕种,造成东北黑土壤的活力退化,也势必改变其中的微生物群落多样性。通过对比原始森林黑土以及良性耕作的菜园黑土的微生物群落,可以认识和了解长期大田耕作黑土微生物群落多样性的改变,对后续的微生物修复具有积极的理论意义。
本试验结果发现,所采集的三江平原大田黑土、菜园黑土和森林黑土三种样本间的微生物群落存在明显差异。大田土的群落丰富度明显低于菜园土和森林土,后两者相似度较高,菜园土的群落丰富度最高,与已报道的趋势一致[13, 16]。通过OTU聚类分析显示,大田土的OTU数比菜园土和森林土分别少13.3%和7.6%。从样本微生物群落组成分析,大田土的微生物群落中,绿弯菌门(Chloroflexi)、浮霉菌门(Planctomycetes)和螺旋体门(Spirochaetes)含量较多,疣微菌门(Verrucomicrobia)、酸杆菌门(Acidobacteria)含量较少,未检测到脱铁杆菌门(Deferribacteres)。森林土中硝化螺旋菌门(Nitrospirae)含量最高,分别约为菜园土和大田土的2倍和4倍,这可能有利于自然界中森林土的自身氮素循环,而大田土和菜园土可能受长期施加不同种类氮素肥料的影响。总体上来看,菜园土的微生物种类最为丰富,这可能跟其种植作物多样性和施用的农家肥种类多样性相关,其中,衣原体门(Chlamydiae)和梭杆菌门(Fusobacteria)的含量明显高于森林土和大田土,这可能跟菜园土施加农家肥引入一些动物源丰富的微生物所致。群落功能基因预测结果也显示出三种土样一些功能基因的差异,其中,大田土的外源性物质降解相关基因相对丰度较高,这可能是由于长期耕种模式下,大田土中产生了一些外源性有害物质,刺激微生物群落中降解相应物质的微生物的含量增加。
东北黑土地因长期的农业耕作发生了变化,也造成了土壤微生物群落和功能的改变。良性的菜园耕种模式对东北黑土可以起到积极的效果,而长期施加化肥的大田耕作模式则起到负面效果。推测施用大田土差异显著的微生物菌群肥料或菌剂,代替或部分代替化学肥料,调节土壤微生物群落结构,将有助于东北耕地黑土的微生物修复,进而提高粮食产量。
参考文献:
[1]高崇升,王建国. 黑土农田土壤有机碳演变研究进展[J]. 中国生态农业学报,2011,19(6): 1468-1474.
[2]魏巍,许艳丽,朱琳,等. 长期施肥对黑土农田土壤微生物群落的影响[J]. 土壤学报,2013,50(2): 372-380.
[3]李东坡,武志杰,陈利军,等. 长期培肥黑土微生物量磷动态变化及影响因素[J]. 应用生态学报,2004,15(10): 1334-1338.
[4]Marschner P, Kandeler E, Marschner B. Structure and function of the soil microbial community in a long-term fertilizer experiment[J]. Soil Biology and Biochemistry, 2003, 35(3): 453-461.
[5]王小兵,吴元元,邓玲. 东北黑土区黑土退化防治与保护研究[J]. 资源与产业,2008,10(3): 81-83.
[6]崔金香,王帅. 土壤微生物多样性研究进展[J]. 河南农业科学,2010(6): 165-169.
[7]王英,王爽,李伟群,等. 长期定位施肥对土壤微生物区系的影响[J]. 东北农业大学学报,2007,38(5): 632-636.
[8]Srivastava S, Singh J,Microbial C. N and P in dry tropical forest soils: effects of alternate land-uses and nutrient flux[J]. Soil Biology and Biochemistry, 1991, 23(2): 117-124.
[9]余悦. 黄河三角洲原生演替中土壤微生物多样性及其与土壤理化性质关系[D]. 济南:山东大学,2012.
[10]张忠民,刘虎,丁延芹,等. 多抗菌剂对植烟土壤细菌群落多样性的影响[J]. 山东农业大学学报(自然科学版),2015(4): 528-532.
[11]樊晓刚,金轲,李兆君,等. 不同施肥和耕作制度下土壤微生物多样性研究进展[J]. 植物营养与肥料学报,2010,16(3): 744-751.
[12]张志明,许艳丽,韩晓增,等. 连续施肥对农田黑土微生物功能多样性的影响[J]. 生态学杂志,2012,31(3): 647-651.
[13]刘晶鑫,迟凤琴,许修宏,等. 长期施肥对农田黑土微生物群落功能多样性的影响[J]. 应用生态学报,2015,26(10): 3066-3072.
[14]Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities[J]. Applied and Environmental Microbiology, 2005, 71(12): 8228-8235.
[15]Langille M G, Zaneveld J, Caporaso J G, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences[J]. Nature Biotechnology, 2013, 31(9): 814-821.
[16]孟庆杰,许艳丽,李春杰,等. 不同施肥/土地利用方式对黑土细菌多样性的影响[J]. 大豆科学,2008,27(3): 480-486.
