一种新型的杀螟丹分析方法及其在稻田杀螟丹残留动态分析中的应用
2016-05-12彭西甜胡西州彭立军
彭西甜, 夏 虹, 张 仙, 胡西州, 彭立军, 沈 菁
(湖北省农业科学院农业质量标准与检测技术研究所/农业部农产品质量安全
风险评估实验室(武汉), 湖北 武汉 430064)
研究论文
一种新型的杀螟丹分析方法及其在稻田杀螟丹残留动态分析中的应用
彭西甜,夏虹,张仙,胡西州,彭立军,沈菁*
(湖北省农业科学院农业质量标准与检测技术研究所/农业部农产品质量安全
风险评估实验室(武汉), 湖北 武汉 430064)
摘要:建立了一种水稻植株、糙米、稻壳、土壤和田水中的杀螟丹气相色谱-火焰光度检测(GC-FPD)方法。样品中的杀螟丹使用稀盐酸提取,然后在碱性条件下使用氯化镍(NiCl2)将其衍生为沙蚕毒素,最后采用在线连接的支撑液液萃取(SLE)和固相萃取(SPE)进行萃取和富集。在优化好的条件下,杀螟丹在5种空白样品中低、中、高3种添加浓度的回收率为80.0%~114.4%,相对标准偏差(RSD)小于13.7%,表明所建立的方法具有良好的准确度和精密度。将所建立的方法用于大田条件下杀螟丹的残留动态分析,为建立杀螟丹的最大残留限量(MRL)提供参考,同时也可对农药施用技术的安全性进行评价。
关键词:支撑液液萃取;气相色谱-火焰光度检测器;杀螟丹;水稻;残留动态
杀螟丹是沙蚕毒素类农药,其在生物体内首先转化为沙蚕毒素,一种从环节动物体内提取出来的神经毒素,该毒素可以与乙酰胆碱受体结合,阻断神经递质的传递,导致害虫神经中枢紊乱,从而起到杀虫的作用[1-3]。目前,杀螟丹作为一种高效的有机杀虫剂,被广泛应用于控制水稻生产中的螟虫和稻纵卷叶螟[4]。然而,有文献报道杀螟丹对一些动物具有一定的毒性[5-7],其在食品和环境中的残留对人体健康造成了威胁。水稻在中国是最重要的食物,其种植面积占到了世界的23%,总产量超过了世界的30%,排在世界第一位[8,9]。因此,发展快速、简单、有效的杀螟丹分析方法,研究杀螟丹在稻田中的消解和残留,对于评价施药技术的安全性,保护环境和人类的健康,具有重要的意义。
目前,光谱法[10]、荧光探针法[11,12]、比色法[13]、气相色谱-电子捕获检测(GC-ECD)[4,14]、液相色谱-质谱法(HPLC-MS)[3,15,16]等已经被用于各种样品中杀螟丹的检测,其中质谱检测方法是目前应用最多的农药残留分析方法。杀螟丹及其衍生物沙蚕毒素分子中含有两个硫原子(见图1),可以通过气相色谱-火焰光度检测(硫)(GC-FPD(S))实现高灵敏、高选择性的分析检测,同时,与HPLC-MS相比,GC-FPD(S)分析成本更低,更容易在常规实验室中得到应用。
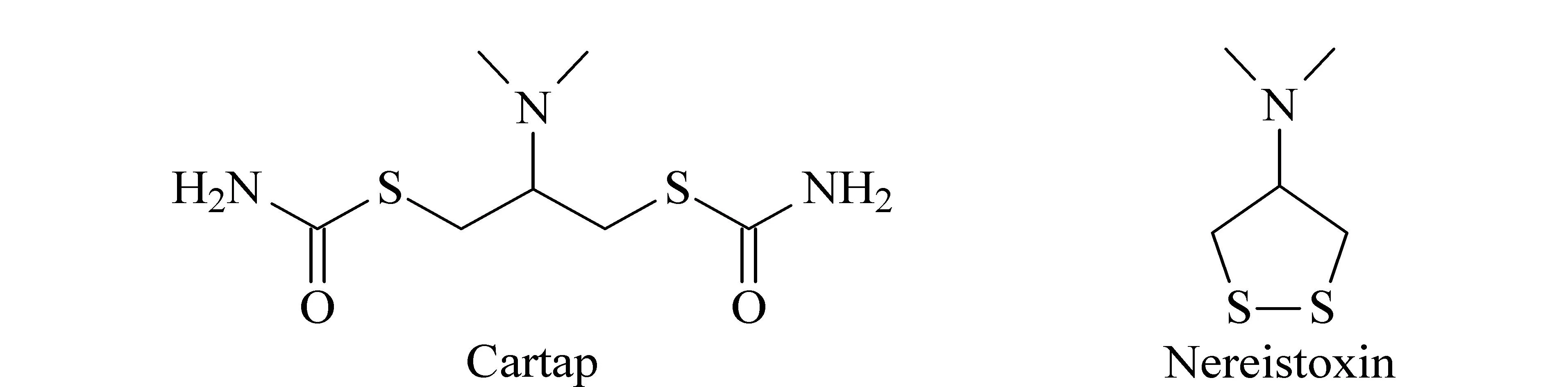
图 1 杀螟丹和沙蚕毒素的化学结构Fig. 1 Chemical structures of cartap and nereistoxin
由于样品基质复杂,杀螟丹在样品中的浓度较低,在仪器分析前进行适当的样品前处理是必不可少的。杀螟丹是一个强极性的碱性化合物,通常通过酸性的水溶液从样品中进行提取,然后采用有机溶剂从碱性水溶液中进行萃取[4]。然而,杀螟丹在碱性条件下不稳定,容易降解为沙蚕毒素,使得分析结果的准确性降低[17,18]。吴刚等[19]在酸性条件下提取茶叶中的杀螟丹,在碱性条件下使用Na2S将杀螟丹衍生为沙蚕毒素,经过乙醚萃取、pH调节、浓缩等步骤,采用GC-FPD(S)分析茶叶中的杀螟丹;该方法需要经过多步的液液萃取(LLE)和浓缩,步骤较为繁琐,不适合大量样品高通量的分析检测;同时,传统的LLE无法避免乳液的形成,有机溶剂的浓缩过程费时费力。支撑液液萃取(SLE)是一种新型的样品前处理技术,由Johnson等[20]在1997年首次提出,其采用一种对水具有强吸附能力、惰性、高比表面积、多孔的固体介质作为载体,以不溶于水的有机溶剂将目标物从载体表面的吸附水层中萃取洗脱下来[21,22],相比于LLE的方法,该方法具有高通量、重复性好、避免形成乳液等优点。
在本文中,我们将SLE和固相萃取(SPE)在线连接用于萃取和富集样品中的杀螟丹衍生物沙蚕毒素,结合GC-FPD(S)的分离检测,建立了一种水稻植株、糙米、稻壳、土壤和田水中杀螟丹快速、高效的分析方法。使用酸性水溶液对样品中的杀螟丹进行提取,在碱性条件下使用氯化镍(NiCl2)将其衍生为沙蚕毒素,然后上硅藻土基的SLE柱,以正己烷洗脱,洗脱液直接采用在线连接的氨基SPE柱进行富集,避免了浓缩过程中分析物的损失,最后使用甲醇将沙蚕毒素从氨基SPE柱上解吸,GC-FPD(S)分离检测。将所建立的方法应用于杀螟丹在水稻稻米、稻壳、植株、土壤、田水中的消解和残留分析。本文的研究结果可以提供杀螟丹在环境中的降解动态数据,为建立杀螟丹的最大残留限量(MRL)提供参考,同时也可对农药施用技术的安全性进行评价。
1实验部分
1.1材料与试剂
杀螟丹盐酸盐(纯度98.1%)购自中国农业部农药检定所(北京)。沙蚕毒素草酸盐(纯度99.0%)购自Dr. Ehrenstorfer GmbH (德国)。0.8%杀螟丹颗粒剂由广东惠州中迅化工有限公司(惠州)提供。HPLC级的甲醇购自T. Baker (荷兰)。分析级的正己烷、盐酸、氨水、二氯化镍(NiCl2)购自国药集团化学试剂有限公司(上海)。Hicapt NH2SPE柱(500 mg, 6 mL)购自维泰克科技有限公司(武汉)。Cleanert@SLE柱(14.5 g, 60 mL)和Cleanert Florisil柱(1.0 g, 6 mL)购自博纳艾杰尔科技有限公司(天津)。
1.2仪器和条件
日本岛津GC-2010, FPD检测器配有S滤光片,毛细管色谱柱Rtx@-35 (300 cm×0.25 mm×0.25 μm) (USA)。程序升温:80 ℃保持2 min,以10 ℃/min升高到200 ℃;不分流进样,进样体积1.0 μL;超纯氮气作为载气,流速为1.0 mL/min;进样口和检测器温度分别为220 ℃和280 ℃。
1.3田间试验
2011年,0.8%的杀螟丹颗粒剂在水稻上消解动态和最终残留的田间试验在湖北省武汉市、浙江省杭州市和河北省石家庄市进行。每个处理包括一个空白对照和3个重复,每小区30 m2。在植株消解动态试验中,0.8%杀螟丹颗粒剂以推荐有效剂量的1.5倍(2 700 g/hm2)分别喷洒在水稻植株表面和田水中,在施药后0.083 (2 h)、1、3、5、7、10、14、21、28、42天采集植株、土壤和田水样品。在最终残留试验中,设高低两个有效剂量(2 700 g/hm2, 1 800 g/hm2),各设施药3次和施药4次,施药间隔期为7天,最后一次施药距采收间隔21天或28天。待水稻成熟时,采集水稻植株、稻穗和土壤样品。
1.4样品制备
首先将水稻植株样品切成1 cm长小段,然后用搅拌器搅拌粉碎。田水样品用塑料瓶在试验点随机采集,分析前过滤。土壤样品在阴凉处晾干后,用搅拌器粉碎,过40目的筛子。水稻样品在室温风干后分离糙米和稻壳,糙米和稻壳样品用搅拌器搅拌成粉末。所有样品分析前都储藏在-20 ℃冰箱中。
1.5样品分析过程
称取一定量的样品(10.0 g的糙米和土壤,5.0 g的植株和稻壳)至100 mL离心管中,加入40 mL的1.0 mol/L盐酸溶液浸泡过夜,超声提取15 min, 4 000 r/min离心5 min,取20 mL上清液至100 mL锥形瓶中,加入1 mL 20 g/L的NiCl2溶液和2.5 mL的氨水,溶液振荡混匀后在室温放置衍生反应1 h,将衍生溶液倒入硅藻土SLE柱,平衡15 min,待样品溶液吸附在SLE柱的硅藻土填料表面后,用60 mL正己烷分3次洗脱硅藻土表面吸附水层中的衍生产物沙蚕毒素,同时洗脱液中的沙蚕毒素采用在线连接的氨基SPE柱进行富集。最后,吸附在氨基SPE柱上的沙蚕毒素采用2.0 mL甲醇进行解吸,GC-FPD(S)分离检测。水样的处理同其他样品处理方法基本相同,将20 mL水样放入100 mL锥形瓶中,后续处理步骤同上。整个分析过程如图2所示。
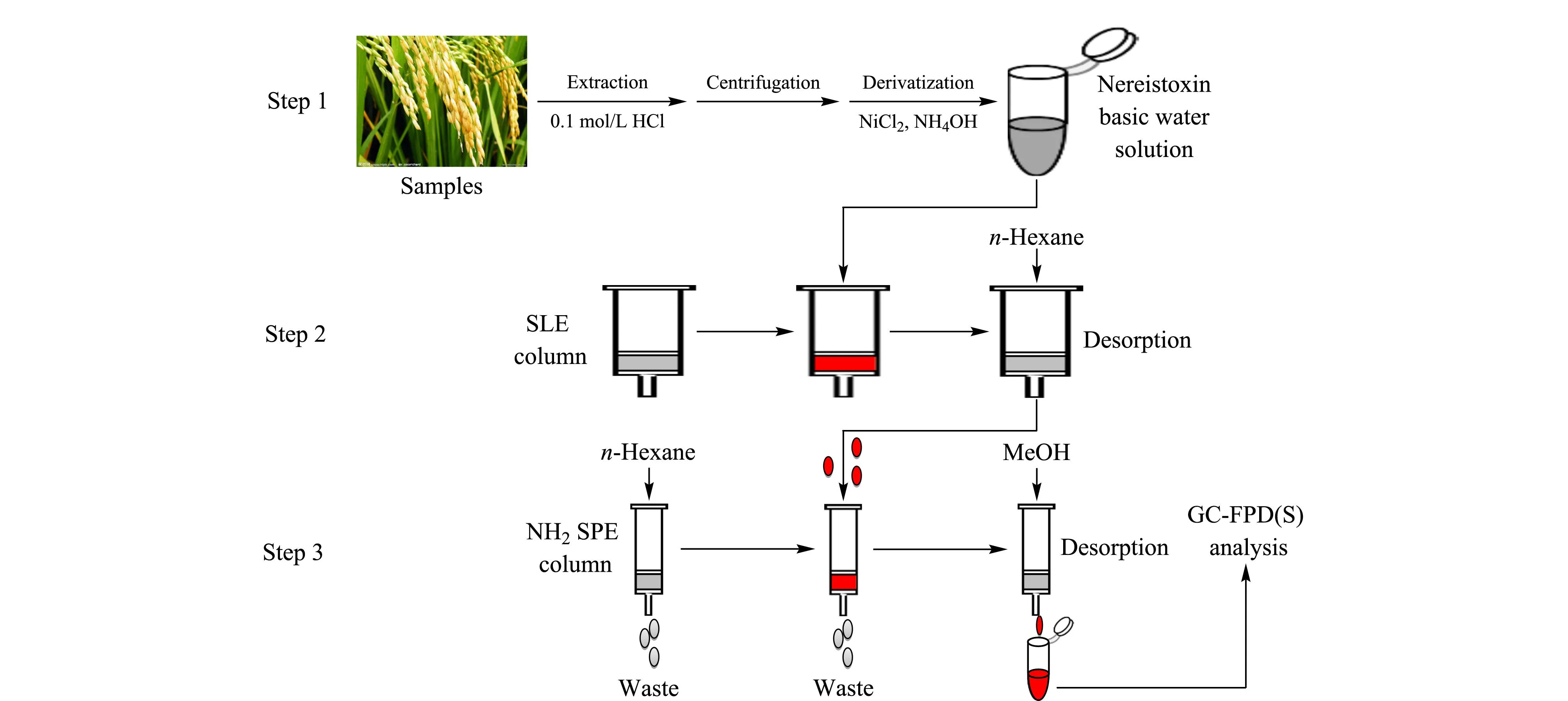
图 2 在线连接SLE-SPE分析植株、糙米、稻壳、土壤和田水中的杀螟丹示意图Fig. 2 Scheme of on-line combination of SLE and SPE for cartap analysis in rice plants, husked rice, rice hull, soil and water
2结果与讨论
2.1样品制备过程的优化
QuEChERS(Quick, Easy, Cheap, Effective, Rugged, Safe)法是目前应用最广泛的食品中农药残留分析的前处理方法[23,24]。在本文中,我们尝试采用传统的QuEChERS方法进行提取,然而由于杀螟丹的水溶性较高,其回收率只有50%左右。此外,我们发现当样品溶液在减压或氮气吹干的条件下进行浓缩时,杀螟丹的衍生物沙蚕毒素很容易挥发,造成回收率降低。
因此,在本文中,通过在线连接SLE和SPE,我们建立了一种水稻植株、糙米、稻壳、土壤和田水中杀螟丹快速、稳定的分析方法。杀螟丹是一种碱性分析物,采用稀盐酸进行提取。对超声提取和振荡提取法进行了比较,发现超声提取15 min回收率基本可以达到100%。然后,使用NiCl2在碱性条件下将杀螟丹衍生为沙蚕毒素,含有沙蚕毒素的样品溶液上硅藻土SLE柱,对3种洗脱溶剂(正己烷、乙醚、乙酸乙酯)的洗脱效率进行了优化,发现60 mL正己烷可以完全将硅藻土表面吸附水层中的沙蚕毒素洗脱下来。最后,采用NH2和Florisil柱在线富集洗脱液中的沙蚕毒素,我们发现两种萃取柱都可以完全萃取洗脱液中的沙蚕毒素。然而,由于强的静电和氢键作用,沙蚕毒素很难从Florisil柱上解吸下来,而2 mL甲醇可以完全解吸氨基柱上的沙蚕毒素。在线的SLE和SPE的结合克服了传统LLE中乳液形成的缺点,避免了浓缩过程中目标物的损失。在最优化的条件下,空白和添加样品的色谱图见图3。

图 3 (a)糙米、(b)稻壳、(c)植株、(d)田水、(e)土壤空白样品和加标样品的GC色谱图Fig. 3 Typical GC chromatograms of blank and spiked samples of (a) husked rice, (b) rice hull, (c) rice plants, (d) water and (e) soil
2.2方法学考察
根据线性范围、定量限(LOQ)、精密度、回收率等对本方法进行了考察。不同浓度的沙蚕毒素标准溶液进入GC-FPD(S)分析。在0.1~40 mg/L的质量浓度范围内,峰面积与分析物的含量具有较好的对数关系,相关方程为lgy=2.04 lgx+6.61,R2为0.999 5,其中y为峰面积,x为分析物的含量(mg/kg)。按照基线与噪声的标准偏差的10倍计算方法的LOQ,糙米和土壤中的LOQ为0.05 mg/kg,植株和稻壳中的LOQ为0.1 mg/kg,田水中的LOQ为0.01 mg/kg。
在空白的植株、糙米、稻壳、土壤和田水样品中添加低、中、高3种浓度的杀螟丹,计算方法回收率和相对标准偏差(RSDs)。回收率是通过计算实测值(采用标准曲线计算)和实际添加值的比得到的。如表1所示,方法回收率为80.0%~114.4%,相对标准偏差小于13.7%,表明所建立的方法具有良好的准确度和精密度,可以用于实际样品的分析检测。
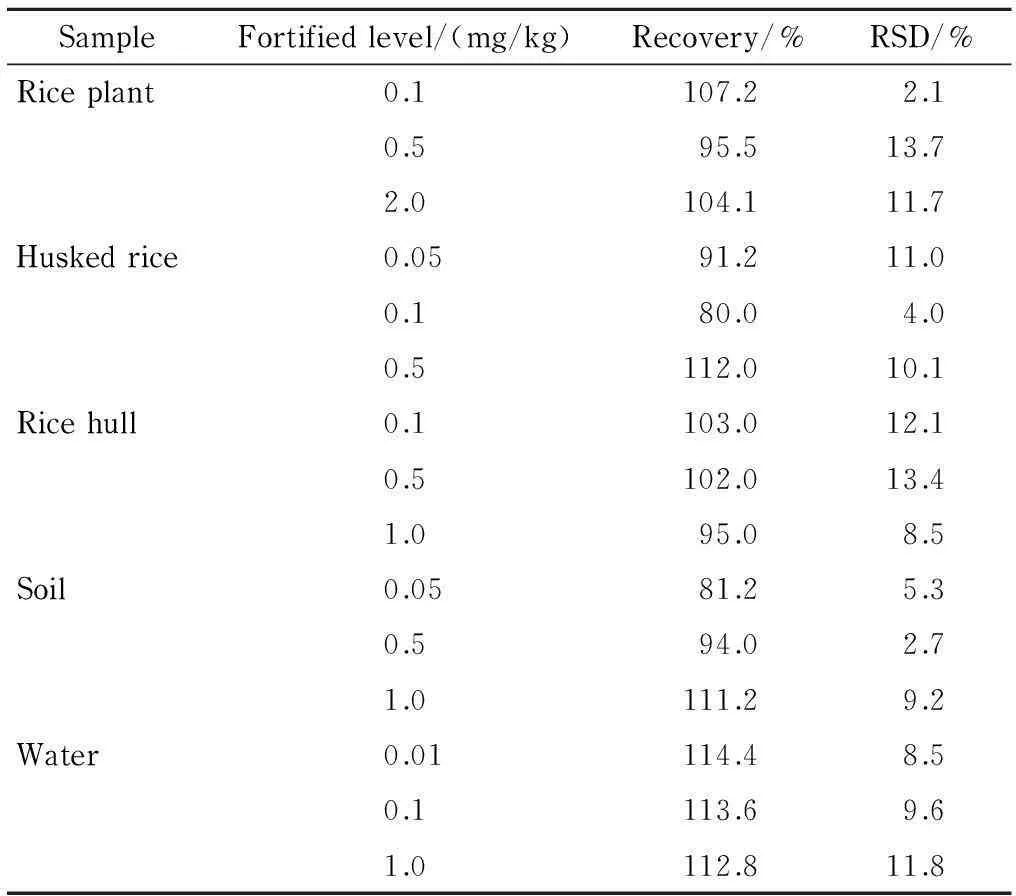
表 1 不同样品中杀螟丹的回收率和相对标准偏差(n=5)
2.3消解试验
稻田水中杀螟丹的消解试验结果如图4a所示,在田水中以2 700 g/hm2的有效剂量施用0.8%杀螟丹颗粒剂后,在三地(湖北、河北、浙江)田水中杀螟丹的初始浓度分别为5.06、3.07和3.73 mg/L,消解动力学可以采用一级动力学方程进行描述:C=5.06e-3.23t(湖北),C=3.08e-1.07t(河北),C=3.72e-1.30t(浙江),其中C和t分别表示质量浓度(mg/L)和时间(天)。根据这3个动力学方程,杀螟丹在湖北、河北、浙江田水中的半衰期(t1/2)分别为0.21、0.65和0.53天,表明杀螟丹在田水中会快速地消解。湖北和浙江水稻植株中杀螟丹的消解曲线如图4b所示,施药3天后杀螟丹在植株中的含量分别为0.12和0.22 mg/kg,在5天后达到峰值,随后快速降低,这可能是由于光照、氧化、水解、生长稀释等原因造成的。在河北植株样品中没有检测到杀螟丹的残留,这可能是气候环境的原因。

图 4 杀螟丹在田水和水稻植株中的消解动态Fig. 4 Dissipation of cartap in paddy water and rice plants
对于杀螟丹在土壤中的消解情况,在河北和湖北的土壤样品中没有检测到杀螟丹的残留,在浙江土壤样品中的检出值也低于0.25 mg/kg,这可能是由于杀螟丹在水中具有很高的溶解度,其消解速度也很快,导致沉积在土壤中的杀螟丹很少。
2.4最终残留试验
将0.8%的杀螟丹颗粒剂以推荐的有效剂量(1 800 g/hm2)和1.5倍的推荐有效剂量(2 700 g/hm2)施药3次或4次(施药间隔期7天)后的第21天和28天后采集样品,杀螟丹在所有的糙米、稻壳、土壤中都没有检出。在水稻植株中的残留量为0.13~1.46 mg/kg,这是由于在水稻生长的后期施用了多次农药,生长稀释的效应较弱,最终导致水稻植株中杀螟丹的检出值较高。
3结论
本文通过在线连接的支撑液液萃取-固相萃取技术,建立了一种高效的水稻植株、糙米、稻壳、土壤、田水中杀螟丹的气相色谱-火焰光度检测(硫)检测方法,方法学考察表明该方法具有良好的准确度和精密度。该方法克服了传统液液萃取的缺陷,避免了样品浓缩过程中分析物的损失。将所建立的方法应用于大田条件下杀螟丹在水稻植株、糙米、稻壳、土壤、田水中的残留动态分析,结果表明在水稻糙米和稻壳中没有检出杀螟丹,杀螟丹在田水和植株中的消解速度很快,田水中的半衰期小于1天,在土壤中的残留量也很低,表明目前的施药方法是安全的。
参考文献:
[1]Lee S J, Tomizawa M, Casida J E. J Agric Food Chem, 2003, 51: 2646
[2]Ferrer C, Mezcua M, Martínez-Uroz, M A, et al. Anal Bioanal Chem, 2010, 398: 2299
[3]Chen H P, Liu X, Wang C P, et al. Journal of Instrumental Analysis, 2013, 32(5): 619
陈红平, 刘新, 王川丕, 等. 分析测试学报, 2013, 32(5): 619
[4]Wu G, Yu H F, Bao X X, et al. Chinese Journal of Chromatography, 2007, 25(2): 288
吴刚, 虞慧芳, 鲍晓霞, 等. 色谱, 2007, 25(2): 288
[5]Zhou S L, Dong Q X, Li S N, et al. Aquat Toxicol, 2009, 95: 339
[6]Liao J W, Kang J J, Jeng C R, et al. Toxicol, 2006, 219: 73
[7]Park Y, Choe S, Lee H, et al. Forensic Sci Int, 2015, 252: 143
[8]Wang K, Wu J X, Zhang H Y. Ecotoxicol Environ Saf, 2012, 86: 111
[9]Fu Y, Zheng Z T, Wei P, et al. Toxicol Environ Chem, 2016, 98(1): 118
[10]Raza N, Aslam M, Noreen Z, et al. Int J Curr Pharm Res, 2013, 5: 54
[11]Jing X, Du L M, Wu H, et al. J Integr Agric, 2012, 11: 1861
[12]Guo J J, Liu X, Gao H T, et al. RSC Adv, 2014, 4(52): 27228
[13]Li W, Zhang D H, Tang Y F, et al. Talanta, 2012, 101: 382
[14]Mahboob S, Al-Ghanim K A, Al-Misned F, et al. Toxicol Environ Chem, 2014, 96(5): 799
[15]Hernando M D, Ferrer C, Ulaszewska M, et al. Anal Bioanal Chem, 2007, 389: 1815
[16]Wang L J, Zhou Y, Huang X Y, et al. Chinese Journal of Chromatography, 2013, 31(12): 1167
王连珠, 周昱, 黄小燕, 等. 色谱, 2013, 31(12): 1167
[17]Asahi Y, Yoshida T. Chem Pharm Bull, 1977, 25: 2211
[18]Lee S J, Caboni P, Tomizawa M, Casida J E. J Agric Food Chem, 2004, 52: 95
[19]Wu G, Yu H F, Bao X X, et al. Journal of Chinese Institute of Food Science and Technology, 2006, 6(5): 129
吴刚, 虞慧芳, 鲍晓霞, 等. 中国食品学报, 2006, 6(5): 129
[20]Johnson C R, Zhang B, Fantauzzi P, et al. Presented at the 5th International Symposium on Solid Phase Synthesis and Combinatorial Chemical Libraries. London, September 1997
[21]Liu H T, Huang L P, Chen Y X, et al. J Chromatogr B, 2015, 992: 96
[22]Bao J M, Ma Z S, Sun Y, et al. Chinese Journal of Chromatography, 2012, 30: 798
包建民, 马志爽, 孙莹, 等. 色谱, 2012, 30: 798
[23]Anastassiades M, Lehotay S J, Stajnbaher D, et al. J AOAC Int, 2003, 86: 412
[24]He Z Y, Wang L, Peng Y, et al. Food Chem, 2015, 169: 372
Development of a novel analytical method of cartap and its application to the residue and dissipation analysis of cartap in paddy field
PENG Xitian, XIA Hong, ZHANG Xian, HU Xizhou, PENG Lijun, SHEN Jing*
(Institute of Agricultural Quality Standards and Testing Technology Research, Hubei Academy of Agricultural Science/Laboratory of Quality & Safety Risk Assessment for Agroproducts (Wuhan), Ministry of Agriculture, Wuhan 430064, China)
Abstract:Cartap has been widely used as a highly effective pesticide in rice fields for the control of rice stem borer and leaf folde. In this paper, a novel analytical method of cartap in rice plants, husked rice, rice hull, soil and water was developed by gas chromatography-flame photometric detector (GC-FPD). Cartap in these sample matrices was first extracted with dilute hydrochloric acid solution, and then derivatized to nereistoxin with nickel chloride (NiCl2) under basic condition. At last, nereistoxin was extracted and concentrated by on-line combination of supported liquid-liquid extraction (SLE) and solid phase extraction (SPE). Under the optimal conditions, the recoveries of cartap in these sample matrices spiked at three levels ranged from 80.0% to 114.4% with relative standard deviations (RSDs) less than 13.7%, indicating good accuracy and precision of our proposed method. The proposed method was then applied to the analysis of residue and dissipation of cartap in rice plants, husked rice, rice hull, soil and water under field conditions. The results can provide reference for the establishment of the maximum residue level (MRL) of cartap and estimation of safety of pesticide application technology.
Key words:supported liquid-liquid extraction (SLE); gas chromatography-flame photometric detector (GC-FPD); cartap; rice; residue and dissipation
中图分类号:O658
文献标识码:A
文章编号:1000-8713(2016)04-0436-06
基金项目:湖北省农科院青年科学基金(2013NKYJJ18);湖北省农业科技创新中心项目(2016-620-000-001-037).
*收稿日期:2016-01-07
DOI:10.3724/SP.J.1123.2015.12034
*通讯联系人.Tel:(027)87389808,E-mail:myshenjing@126.com.
Foundation item: Youth Fund for Hubei Academy of Agricultural Science (No. 2013NKYJJ18); Project of Hubei Provincial Innovation Center for Agricultural Sciences and Technologies (No. 2016-620-000-001-037).
