酒精性肝病大鼠肝组织PPARα表达及意义
2016-05-10曹智丽周俊英王娟王艳王维张文双
曹智丽,周俊英,王娟,王艳,王维,张文双
(1河北北方学院附属第二医院,河北张家口075100;2河北医科大学第三医院)
酒精性肝病大鼠肝组织PPARα表达及意义
曹智丽1,周俊英2,王娟1,王艳1,王维1,张文双1
(1河北北方学院附属第二医院,河北张家口075100;2河北医科大学第三医院)
目的 探讨过氧化物酶体增殖物激活受体α(PPARα)在酒精性肝病发生、发展中的作用。方法 选择Wistar大鼠50只,随机取40只采用乙醇灌胃法制备酒精性肝病模型,根据造模时间不同分为模型4、8、12、16周组。至造模结束,共成模35只,模型4、8、12、16周组分别10、9、8、8只;剩余10只作为正常对照组,不予任何处理。各组处死后留取肝组织标本。采用免疫组化染色法、Western blotting法、RT-qPCR法检测PPARα蛋白及mRNA表达。结果 免疫组化染色显示,PPARα蛋白定位于肝细胞胞质,呈棕黄色染色。正常对照组肝细胞PPARα蛋白表达丰富;随着造模时间的延长,各模型组肝组织PPARα蛋白表达逐渐减少,模型16周组肝组织仅见少量PPARα蛋白表达。Western blotting、qRT-PCR检测结果显示,各模型组PPARα蛋白及mRNA表达均低于正常对照组(P均<0.05);随着造模时间的延长,PPARα蛋白及mRNA表达逐渐降低(P均<0.05)。结论 PPARα可能参与酒精性肝病的发生、发展。
酒精性肝病;过氧化物酶体增殖物激活受体α;大鼠
近年来,酒精性肝病的发病率逐年升高,已成为继病毒性肝炎后导致肝损伤的第二大病因。酒精性肝病的病理变化最初主要为肝细胞脂肪变性,随后进展为脂肪性肝炎、肝纤维化,最终导致肝硬化。过氧化物酶体增殖物激活受体(PPARs)是核激素受体家族中的配体激活受体,分为α、β、γ三种亚型,其中PPARα可通过多种途径参与脂质代谢的调节。但其在酒精性肝病发病过程中作用的研究鲜见报道。2013年12月~2014年12月,我们观察了酒精性肝病大鼠肝组织PPARα表达,现分析结果并探讨其在酒精性肝病发生、发展中的作用。
1 材料与方法
1.1 材料 健康雄性Wistar大鼠50只,清洁级,3月龄,体质量(200±20)g,由河北医科大学动物实验中心提供,动物许可证号:SCXK(冀)2008-1-003。饲养环境:温度(22±2)℃,湿度(70±5)%。主要仪器:UV-2550PCX型紫外分光光度计,宁波新芝生物科技股份有限公司;ABI7500型实时荧光定量PCR仪,美国ABI公司;电泳仪,美国Hoefer公司;5417R型台式高速冷冻离心机,德国EP公司。主要试剂:TRIzol试剂,北京康为世纪生物科技有限公司;PCR逆转录试剂盒、荧光定量PCR试剂盒、PPARα上下游引物、β-actin上下游引物,美国Invitrogen公司;PPARα抗体,美国Epitomics公司;β-actin抗体,杭州华安生物有限公司;辣根过氧化物酶标记的羊抗兔二抗,美国KPL公司;ECL发光液,美国Santa Cruz公司;RIPA细胞裂解液,北京索莱宝科技有限公司;PVDF膜,美国Millpore公司。
1.2 模型制备及干预 所有大鼠适应性喂养1周,随机取40只采用乙醇灌胃法[1]建立酒精性肝病模型。根据造模时间不同分为模型4、8、12、16周组,每组10只。1~4周给予30%乙醇5.0 g/(kg·d)灌胃,5~8周给予40%乙醇6.0 g/(kg·d)灌胃,9~12周给予50%乙醇7.0 g/(kg·d)灌胃,13~16周给予60%乙醇8.0 g/(kg·d)灌胃。每日9:00灌胃,1次/d。模型制备过程中5只大鼠因乙醇误吸入气管窒息死亡,模型4、8、12、16周组分别有10、9、8、8只成模。各组造模结束禁食12 h处死,留取肝组织标本。剩余10只作为正常对照组,不予任何处理,直接处死留取肝组织标本。
1.3 相关指标观察
1.3.1 肝组织PPARα蛋白表达 ①采用免疫组化法。取各组肝组织,4%中性甲醛固定,石蜡包埋,常规方法制备组织切片,免疫组化染色,显微镜下观察。②采用Western blotting法。取100 mg肝组织制备组织匀浆;提取蛋白并定量后,行SDS-PAGE电泳,蛋白转移至PVDF膜;将PPARα、β-actin一抗加入不同标记的PVDF膜上,洗膜、定影,最后ECL显影。应用Image J软件测定条带灰度值,以目标条带与内参条带灰度的比值作为PPARα蛋白的相对表达量。
1.3.2 肝组织PPARα mRNA表达 采用RT-qPCR法。取部分肝组织以TRIzol法提取RNA,逆转录为cDNA,应用荧光定量PCR试剂盒、ABI7500型实时荧光定量PCR仪测定PPARα mRNA表达。引物由美国Invitrogen公司合成,PPARα:上游引物5′-CACGATGCTGTCCTCCTT-3′,下游引物5′-TCCAGTTCGAGGGCATTG-3′;β-actin:上游引物5′-CCATGTACGTAGCCATCC-3′,下游引物5′-CTCAGCTGTGGTGGTGAA-3′。PCR反应条件:95 ℃预变性2 min,95 ℃变性15 s,60 ℃退火30 s,72 ℃延伸45 s,共40个循环。以2-ΔΔCt法计算PPARα mRNA的相对表达量。

2 结果
免疫组化染色显示,PPARα蛋白定位于肝细胞胞质,呈棕黄色染色。正常对照组肝细胞PPARα蛋白表达丰富(插页Ⅱ图5A);随着造模时间延长,各模型组肝组织PPARα蛋白表达逐渐减少(插页Ⅱ图5B、5C、5D),至模型16周组时肝组织仅见少量PPARα蛋白表达(插页Ⅱ图5E)。
正常对照组肝组织PPARα蛋白及mRNA相对表达量均高于各模型组(P均<0.05)。随着造模时间的延长,各模型组肝组织PPARα蛋白及mRNA相对表达量逐渐降低(P均<0.05)。各组肝组织PPARα蛋白及mRNA相对表达量比较见表1。
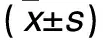
表1 各组肝组织PPARα蛋白及mRNA相对表达量比较
注:与正常对照组比较,*P<0.05;与模型4周组比较,#P<0.05;与模型8周组比较,△P<0.05;与模型12周组比较,▲P<0.05。
3 讨论
酒精性肝病是由于长期大量饮酒导致的肝脏疾病,随着病程进展,可逐渐发展为酒精性脂肪肝、酒精性肝炎、酒精性肝硬化等[2]。酒精性肝病早期典型的病理变化是肝细胞脂肪变性,一旦去除乙醇诱因,脂肪变性往往能够恢复正常。若脂肪合成、分解、代谢等出现异常,代谢紊乱会加重肝细胞损伤,且肝细胞损伤与肝脏脂肪沉积的程度密切相关。在乙醇引起肝细胞脂肪沉积的基础上肝细胞受到氧化应激作用,炎症反应加重,可进一步加重肝脏的病理损伤[3,4]。
PPARα是一种参与调节脂肪酸氧化和运输的核激素受体,其参与脂质代谢的各个环节,包括脂肪酸在细胞内结合、脂肪酸透膜吸收、脂蛋白合成与转运等,其表达与肝脏乙醇代谢和脂质代谢密切相关[5]。有研究证实,乙醇通过抑制PPARα的表达及破坏PPARα蛋白的功能,干预脂肪酸的氧化及转运途径,抑制甘油三酯合成,最终引起脂质代谢障碍[6];而脂质代谢失衡可导致肝内脂质聚集过多,超过肝负荷,即引起肝细胞脂肪变性,肝组织内过量脂质浸润可引起线粒体损伤、氧化应激及炎症反应,进一步加重肝损伤[7]。Lebrun等[8]研究发现,随着酒精性肝病造模时间延长,肝组织脂肪沉积逐渐增多,至造模结束时肝细胞内已呈弥漫小泡性脂肪变性。Nakajima等[9]研究发现,乙醇长期喂养可降低小鼠肝组织PPARα mRNA和蛋白的表达,下调PPARα的靶基因表达,如酰基辅酶α氧化酶、中链酰基辅酶α脱氢酶、脂肪酸结合蛋白等;有研究还发现,应用乙醇诱导PPARα基因敲除的小鼠比野生型小鼠更易发生肝损伤,说明PPARα表达可被乙醇抑制,从而导致脂肪酸β氧化减少,促进脂肪酸和甘油三酯的合成,进一步导致肝细胞脂肪变性的发生[10~14]。Sahebkar等[15]研究也证实,随着乙醇代谢产物乙醛生成量的增多,PPARα表达逐渐减少,肝细胞所受伤害和毒性作用相继出现,如线粒体损伤、炎性损伤等。
本研究各模型组PPARα蛋白及mRNA表达均低于正常对照组;随着造模时间延长,各模型组肝组织PPARα蛋白及mRNA表达逐渐降低。推测激活PPARα受体可促进脂肪酸氧化基因、脂质合成基因的表达,从而改善乙醇引起的脂质代谢紊乱,减轻肝细胞脂肪累积[16~19]。
综上所述,我们认为PPARα可能参与酒精性肝病的发生、发展,其具体作用机制尚不清楚,仍需进一步研究。
[1] 周俊英,周东方,王玮.酒精直接灌胃法建立大鼠酒精性肝病模型[J].河北医科大学学报,2009,30(4):328-330.
[2] 中华医学会肝病学分会脂肪肝和酒精性肝病学组.酒精性肝病诊疗指南(2010年修订版)[J].中华肝脏病杂志,2010,18(3):167-170.
[3] Keller JM, Collet P, Bianchi A, et al. Implications of peroxisome proliferator-activated receptors(PPARS)in development, cell life status and disease[J]. Int J Dev Biol, 2000,44(5):429-442.
[4] Kong L, Ren W, Li W, et al. Activation of peroxisome proliferator activated receptor alpha ameliorates ethanol induced steatohepatitis in mice[J]. Lipids Health Dis, 2011(10):246.
[5] Konig B, Koch A, Spielmann J, et al. Activation of PPAR alpha lowers synthesis and concentration of cholesterol by reduction of nuclear SREBP-2[J]. Biochem Pharmacol, 2007,73(4):574-585.
[6] 杨辉,李瑜元,聂玉强,等.过氧化物酶体增殖因子活化受体-基因多态性与非酒精性脂肪性肝病及脂联素的关系[J].中华消化杂志,2008,28(2):113-115.
[7] Nan YM, Kong LB, Ren WG, et al. Activation of peroxisome proliferator activated receptorα ameliorates ethanol mediated 1iver fibrosis in mice[J]. Lipids Health Dis, 2013(12):21.
[8] Lebrun V, Molendi-Coste O, Lanthier N, et al. Impact of PPAR-α induction on glucose homoeostasis in alcohol-fed mice[J]. Clin Sci(Lond), 2013,125(11):501-511.
[9] Nakajima T, Kamijo Y, Tanaka N, et al. Peroxisome proliferator-activated receptor a protects against alcohol-induced liver damage[J]. Hepatology, 2004,40(4):972-980.
[10] 陆璐,钱建成,胡晨波,等.过氧化物酶体增殖物激活受体γC161-T、细胞色素P450ⅡE1基因多态性与脂肪性肝病相关性研究[J].中国医刊,2013,48(6):46-49.
[11] 王玮,周俊英,赵彩彦,等.脂联素及其受体在酒精性肝病大鼠肝组织中的表达下降[J].基础医学与临床,2010,30(2):170-174.
[12] 许新,姜曼,李江文,等.过氧化物酶体增殖物激活受体γ基因rs3856806位点多态性与非酒精性脂肪性肝病的相关性分析[J].临床肝胆病杂志,2015,31(7):1088-1091.
[13] 姜潇,白晓彦,伍会健.PPARs在非酒精性脂肪性肝病发病机制中的作用[J].生理科学进展,2015,46(3):220-224.
[14] 金文君.过氧化物酶体增殖物激活受体与脂肪性肝病的关系[J].实验与检验医学,2008,26(1):65-66.
[15] Sahebkar A, Chew GT, Watts GF. New peroxisome proliferator-activated receptor agonists: potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease[J]. Exper Opin Pharmacother, 2014,15(4):493-503.
[16] GaIli A, Pinaire J, Fischer M, et al. The transcrip tional and DNA binding activity of peroxisome proliferator-activated receptor alpha is inhibited by ethanol metabolism. A nove1 mechanism for the development of ethanol-induced fatty liver[J]. J Biol Chem, 2001,276(1):68-75.
[17] 陶妍,吴高峰,杨建成,等.PPAR-α在酒精性脂肪肝发生中的作用研究进展[J].动物医学进展,2012,32(2):71-74.
[18] 郭美祥,田晓娟,李文彪.过氧化物酶体增殖物激活受体与酒精性肝病关系研究进展[J].中西医结合肝病杂志,2006,16(5):306-308.
[19] 黎玉.酒精性脂肪肝分子机制的研究进展[J].中华肝脏病杂志,2006,14(11):878-880.
张家口市科技计划自筹经费项目(1521060D)。
周俊英(E-mail: doctorzhoujy@163.com)
10.3969/j.issn.1002-266X.2016.24.010
R575.5
A
1002-266X(2016)24-0031-03
2016-01-12)
