米枯力兹病51例临床病理分析
2016-05-06翟晓利楼善贤施红旗胡斌章艳斐金华市中心医院病理科浙江金华321000
翟晓利,楼善贤,施红旗,胡斌,章艳斐(金华市中心医院 病理科,浙江 金华 321000)
米枯力兹病51例临床病理分析
翟晓利,楼善贤,施红旗,胡斌,章艳斐
(金华市中心医院病理科,浙江金华321000)
[摘 要]目的:探讨米枯力兹病的病理诊断、鉴别诊断及预后等问题,为临床防治提供依据。方法:对51例米枯力兹病的临床和病理资料进行分析,并进行LCA、CD20、CD3、CD4、CD45RA、CD45RO、EMA、CEA、S-100、CK、lysozyme、EBV、IgG4、HBsAg、HBcAg免疫组织化学标记。结果:米枯力兹病的病变特征为腺实质萎缩和淋巴样组织增生。免疫组织化学标记示T和B淋巴细胞混合性增生,IgG4 80%阳性。结论:米枯力兹病为一种自身免疫性疾病,多见于绝经后女性,手术后可以复发,治疗上应辅以免疫调节剂等治疗。
[关键词]米枯力兹病;自身免疫性疾病;免疫组织化学标记
米枯力兹病(Mikulicz’s disease),又称良性淋巴上皮病变,是一种表现为慢性、无痛性、双侧或单侧腮腺、颌下腺及泪腺肿大,以腺实质萎缩和淋巴细胞反应性增生为特征的自身免疫性疾病[1]。现在认为,此病并非一个独立的疾病,而是斯耶格伦(Sjogrn)综合征的一种表现。该疾病的真正原因尚未阐明,系炎性或肿瘤性病变尚无定论。现将我院近17年来收治的51例米枯力兹病进行分析,报告如下。
1 资料和方法
1.1一般资料 收集1997年至2014年入院治疗的51例米枯力兹病患者,其中男15例,女36例,男女比例为1:2.4;年龄19~72岁,其中40岁以上42例(占82.4%),50岁以上绝经后女性30例(占59%);发生于腮腺24例,颌下腺18例,腭部2例,泪腺2例,舌下腺5例。病程平均1~3个月,最长1年。绝大部分表现为无痛性肿块,多呈单侧唾液腺肿大,6例血沉加快,3例有典型斯耶格伦综合征表现。10例有复发史,其中2例复发3次以上。
1.2方法 51例患者的手术切除标本,经4%甲醛固定,常规脱水,石蜡包埋,4 μm切片;常规HE染色;光镜观察。并作苦味酸-酸性品红法(van gieson,VG)、银氨染色法(gordon sweets,GS)、阿尔辛蓝(Alcian blue,AB)染色法特殊染色,观察胶原纤维、网状纤维、黏液分布情况。采用Envision二步法,选择LCA(克隆号PD7/26+2B11)、CD20(克隆号L26)、CD3(克隆号SP7)、CD4(克隆号SP35)、CD45RA(克隆号111-1C5)、CD45RO(克隆号UCHL-1)、EMA(克隆号E29)、CEA(克隆号ZC23)、S-100(克隆号L26)、CK19(克隆号A53-B/A2.26)、lysozyme(多克隆)、EBV(克隆号CS1-4)、IgG4(克隆号HP6025)、HBsAg(克隆号3E7)、HBcAg(多克隆),对标本进行免疫组织化学标记。对有关临床和病理资料进行统计分析处理。抗体和试剂盒购自福州迈新生物技术开发有限公司。
2 结果
2.1巨检 病变腺体多呈弥漫性肿大,小叶结构清晰可见,少数呈局限性肿块,分界清楚,无或有不明显的包膜。切面灰白或灰黄色,质韧有弹性,部分可见小囊腔,肿块直径1~4 cm。
2.2镜检 HE染色结果示:淋巴样组织增生,大部分腺实质萎缩消失,每个淋巴组织浸润灶中淋巴细胞均达到50个以上[2]。根据淋巴组织增生及腺泡、导管萎缩情况,本研究认为可将米枯力兹病分三级,一级:轻度病变即腺泡导管萎缩减少、淋巴样组织增生,两者各占50%;二级:中度病变,淋巴样组织约占视野2/3左右,腺泡、闰管等明显减少,腺泡甚或消失,萎缩腺泡可形成上皮岛样结构,淋巴细胞密集分布,可见少量滤泡形成;三级:重度病变,淋巴样组织几乎占据全部视野,仅在个别区域存在少量腺泡、闰管,腺泡甚至消失,萎缩腺泡多形成上皮岛样结构,淋巴组织中见巨噬细胞、滤泡树突状细胞明显增生,多个滤泡形成(见图1)[4]。本组资料轻度病变24例,中度17例,重度10例,重度病变均为复发病例。
2.3特殊染色 病变边缘可见VG染色,提示胶原纤维增生。GS染色见网状纤维分布正常。AB染色未见黏液成分。
2.4免疫组织化学标记 淋巴样组织LCA阳性,CD20、CD45RA阳性呈散在或灶状分布,CD3、CD4多呈散在阳性,提示米枯力兹病T、B淋巴细胞均增生,但以B淋巴细胞增生为主(见图2)。残存的腺泡、闰管等可见EMA、CK19阳性(见图3)或弱阳性,CEA阴性。腺泡、闰管周围可见S-100阳性的肌上皮细胞。增生的淋巴组织中见S-100阳性的树枝状网状细胞和溶菌酶阳性的组织细胞,在重度病变的病例中,这2种细胞增生明显。80%病例免疫组织化学标记IgG4抗体表达阳性,主要表达在浆细胞的胞浆中(见图4)。

图1 腮腺组织HE染色示淋巴组织增生,腺体萎缩改变(×100)

图2 腮腺组织免疫组织化学染色CD 20结果显示强阳性,提示以B淋巴细胞增生为主(×100)

图3 腮腺组织免疫组织化学染色CK19强阳性,显示残余腺体及上皮岛(×100)
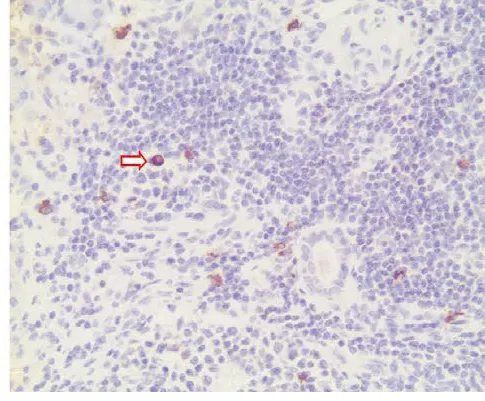
图4 腮腺组织免疫组织化学染色IgG4阳性,显示浆细胞增生(×200)
3 讨论
3.1病因及病理诊断 米枯力兹病多发生于中老年女性,尤其以绝经后女性多见。本组女性为男性的2.4倍,40岁以上占82.4%,50岁以上绝经后女性占59%,这与之前文献[2]报道一致。发生部位多为腮腺,其次为颌下腺。一般认为米枯力兹病是一种自身免疫性疾病。本组80%病例IgG4阳性,3例有典型斯耶格伦综合征表现,提示该病与自身免疫功能紊乱有关。斯耶格伦综合征主要以干性角膜结膜炎、类风湿性关节炎和腮腺肿大为特征,其腮腺病变特征与米枯力兹病无区别,二者均为涎腺组织内淋巴细胞增生性病变,但米枯力兹病主要是局限于涎腺和泪腺,而斯耶格伦综合征有更多的全身变化,米枯力兹病是斯耶格伦综合征的一种变异,或不完全型[2-8]。由于老年人免疫功能低下,易患自身免疫性疾病,这也与米枯力兹病多见于老年人有关。病变特征为淋巴细胞增生,尤以B淋巴细胞增生为主,并形成多量浆细胞及产生IgG4抗体,良性淋巴上皮病变中镜下IgG4阳性细胞大于50%,每个高倍视野中浆细胞数大于10%。EB病毒、HBsAg、HBcAg表达阴性,符合自身免疫性疾病特征;并见巨噬细胞、滤泡树突状细胞增生,而腺泡、闰管则萎缩,也符合免疫病理的改变。随着疾病的发展,淋巴组织反应性增生,导致腺泡、闰管的萎缩,并形成上皮肌上皮岛[4],后者代表退化过程中萎陷的腺泡,主要由上皮细胞和肌上皮细胞所构成,导管上皮细胞则极少参与[5],由于泪腺、腮腺、颌下腺的萎缩,故患者出现口干、眼干诸症。米枯力兹病的诊断主要依据临床表现、光镜下见淋巴组织反应性增生、腺泡和导管上皮萎缩等病理特征,结合免疫组化为T、B淋巴细胞混合性增生,同时还有巨噬细胞、滤泡树突状细胞的增生等即可作出病理诊断。
3.2鉴别诊断及预后探讨 米枯力兹病病理改变主要应与慢性诞腺炎鉴别,后者病理切片背景中可见多种炎症细胞成分,包括淋巴细胞、中性粒细胞、嗜酸性粒细胞等,并见纤维组织增生、胶原化病变。由于纤维化使腺泡、导管数量相对减少。重度的米枯力兹病还应与恶性淋巴瘤、淋巴上皮癌鉴别,免疫组织化学标记见淋巴细胞单克隆性增生有助于恶性淋巴瘤的诊断,上皮组织的免疫组织化学标记及周围浸润情况也有助于淋巴上皮癌的诊断。米枯力兹病还应与唾液腺的其他疾病如乳头状淋巴细胞性囊腺瘤、恶性淋巴瘤、转移性癌等鉴别[4]。明确诊断及病因分析有利于对米枯力兹病的进一步防治。治疗上除了手术切除肿块外,应强调免疫功能的调节及免疫治疗。由于米枯力兹病为自身免疫性疾病,治疗上较为困难,手术后也常有复发,复发后病变比前次更为严重,与恶性肿瘤症状上难以区别,但米枯力兹病无异形性,也无浸润、转移。疾病发展过程中,淋巴组织可能发生恶变成为淋巴瘤,而上皮成分发生恶变则成为淋巴上皮癌[6]。有些病例可伴有其他系统的恶性肿瘤发生[7,9-10]。
参考文献:
[1]刘彤华.诊断病理学[M].3版.北京:人民卫生出版社,2013:19 -20.
[2]武忠弼.中华外科病理学[M].北京:人民卫生出版社,2002:517 -519.
[3]罗塞.阿克曼外科病理学[M].10版.郑杰,译.北京:北京大学出版社,2014:820 -821.
[4]同济医科大学病理学教研室,中山医科大学教研室.外科病理学[M].2版.武汉:湖北科学技术出版社,1999:69 -70.
[5]CHAUDHRY A P,CUTLER L S,YAMANE G M,et al.Light and ultrastructural features of lymphoepithelial lesions of the salivary glands in Mikulicz’s disease[J].J Pathol,1986,148(3):239-250.
[6]GRAVANIS M B,GIANSANTI J S.Malignant histopathologic counterpart of the benign lymphoepithelial lesion[J].Cancer,1970,26(6):1332-1342.
[7]PINKUS G S,DEKKER A.Benign lymphoepithelial lesions of the parotid glands associated with reticulum cell sarcoma.Report of a case and revien of the literature[J].Cancer,1970,25(1):121-127.
[8]WU Y,XU Z R,ZHOU W J,et al.Immunoglobulin G4-related disease with features of Mikullicz’s disease and autoimmune pcmcreatitis which firstly presented as asymptomatic lymphadenopathy:a case report[J].Chin Med J (Engl),2015,128(5):706-707.
[9]MORIYAMA M,FURUKAWA S,KAWANO S,et al.The diagnostic utility of biopsies from the submanclibular and labial Salivary glands in IgG4-revated dacryoaclenitis’s and sialoaclenitis,so-called mikulicz’s disease[J].J oral Maxillofac Surg,2014,43(10):1276-1281.
[10]FURUKAWA S,MORIYAMA M,TANAKA A,et al.Preferential M2 macrophages contribute to fibrosis in IgG4-related dacryoaclenitis and sialoaclenitis,so-called Mikulicz’s disease[J].Clin Immunol,2015,156(1):9-18.
(本文编辑:赵翠翠,丁敏娇)
·临 床 经 验·
Mikulicz’s disease:a clinicopathological analysis of51 cases
ZHAI Xiaoli1,LOU Shanxian2,SHI Hongqi2,HU Bin2,ZHANG Yanfei2.Department of Pathology,Jinhua Municipal Hospital,Jinhua,321000
Abstract:Objective:To explore the pathological features,differential diagnosis and prognosis of Mikulicz’s disease,and provide methods of treatment and prevention for clinicians.Methods:The clinical and pathological features of Mikulicz disease were analyzed in 51 patients and the expression of LCA,CD 20,CD 3,CD 4,CD 45RA,CD 45RO,EMA,CEA,S-100,CK,lysozyme,EBV,IgG4,HBsAg and HBcAg in Mikulicz disease were detected immunohistochemically.Results:Microscopically,the prominent features of Mikulicz disease were acinar atrophy and lymphoid tissue hyperplasia.Immunohistochemical staining showed that the lymphoid tissue was composed of a mixed population of B and T lymphocytes and about 80% of patients were IgG4 positive.Conclusion:Mikulicz disease is an autoimmune disease and mostly occurred in postmenopausal women.Postoperative recurrence may happen.Immunosuppressive agents may be helpful in treatment.
Key words:Mikulicz’s disease; autoimmune disease; immuno histochemistry
作者简介:翟晓利(1978-),女,河南新乡人,主治医师。
收稿日期:2015-04-03
[中图分类号]R365
[文献标志码]B
DOI:10.3969/j.issn.2095-9400.2016.03.018
