MBNER:面向生物医学领域的多种实体识别系统
2016-05-03杨志豪林鸿飞宫本东
杨 娅,杨志豪,林鸿飞,宫本东,王 健
(1.大连理工大学 计算机科学与技术学院,辽宁 大连 116024;2. 中国烟草总公司大连市公司,辽宁 大连 116010)
MBNER:面向生物医学领域的多种实体识别系统
杨 娅1,杨志豪1,林鸿飞1,宫本东2,王 健1
(1.大连理工大学 计算机科学与技术学院,辽宁 大连 116024;2. 中国烟草总公司大连市公司,辽宁 大连 116010)
生物命名实体识别,就是从生物医学文本中识别出指定类型的名称。目前,面向生物医学领域的实体识别研究不断出现,从海量生物医学文本自动提取生物实体信息的技术变得尤为重要。该文介绍了一个面向生物医学领域的多实体识别系统MBNER(Multiple Biomedical Named Entity Recognizer)。该系统可以在生物医学文本中同时识别出基因(蛋白质)、药物、疾病实体,其对基因(蛋白质)、药物、疾病实体识别在各自数据集上分别得到了89.05%,76.73%,90.12%的综合分类率(F-score)。该系统以可视化的形式给出对三种命名实体的识别结果。
机器学习;特征耦合泛化;CRF;全称缩写对
1 引言
文本挖掘是指从大量文本数据中抽取事先未知的、可理解的、最终可用的知识的过程,同时运用这些知识更好地组织信息以供将来参考[1]。生物医学研究是目前最热门的研究领域之一,发表在该领域的文献平均每年以60万篇的速度增长,人工挖掘费时费力。因此如何从海量的生物医学文本中自动挖掘出有价值的信息,是该领域研究者所面临的挑战。生物医学文本挖掘,作为生物信息学的分支之一,应运而生并取得了重大的进展。利用生物医学文本挖掘,可以在多方面辅助生物医学研究者的工作,大大提高文本挖掘的效率。
生物医学文本挖掘的基本任务之一就是生物医学文本中命名实体的识别[2]。生物命名实体识别,就是从生物医学文本中识别出指定类型的名称,比如基因、蛋白质、核糖核酸、脱氧核糖核酸、疾病、细胞、药物的名称等[3]。由于生物医学文献的规模庞大,各种专有名词不断涌现,一个专有名词往往有很多同义词,而且普遍存在大量的缩写词,人工识别费时费力,因此如何对命名实体进行识别就变得尤为重要。命名实体识别是文本挖掘系统中的一个重要的基础步骤,命名实体识别的准确程度是其他文本挖掘技术如信息提取或文本分类等的先决条件。郑强等[4]介绍了目前命名实体识别的特点,并从评测方法、特征选择、机器学习方法和后期处理等方面综述了生物医学命名实体识别的研究方法、成果、存在的问题及可能的研究方向。得出的结论是近年来生物医学命名实体识别在语料库、特征选择、识别方法等方面取得了一定的进展,但由于生物医学领域的命名实体具有一些独特的特点,要使系统达到更高的性能仍面临着很大的挑战。
目前,使用比较多的生物命名实体识别的研究方法主要有以下几种:基于规则的方法[5]、词典匹配的方法[6]以及机器学习的方法,如支持向量机(SVM)[7]、最大熵[8]、条件随机场(CRF)[9]以及隐马尔科夫(HMM)[10]等。目前,大部分生物命名实体相关的研究只针对一种命名实体进行识别,如对基因(蛋白质)的识别研究,对药物的识别研究等。
本文介绍了一个面向生物医学领域的多实体识别系统MBNER(Multiple Biomedical Named Entity Recognizer) ,可以在生物医学文本中同时识别出基因(蛋白质)、药物、疾病实体。在基因(蛋白质)命名实体识别过程中,首先利用生物医学相关的数据库生成词典, 然后用基于特征耦合泛化(Feature Coupling Generalization)的半监督学习方法[11]对词典去噪, 最后用词典结合CRF的方法对基因(蛋白质)命名实体进行识别;在疾病实体识别过程中,使用词典结合CRF的方法对疾病命名实体进行识别,然后利用全称缩写词对对标注结果进行后处理;在药物实体识别过程中,从DRUGBANK中抽取药名作为种子,然后从PUBMED摘要中提取模板,用生成的模板再到PUBMED摘要中提取药名,从而生成初步的药名词典。然后用基于特征耦合泛化的学习方法对词典去噪,接着用词典结合CRF的方法对药物命名实体进行识别。
2 方法介绍
2.1 系统框架图
该系统的框架图如图1所示。该框架图的处理流程如下:
(1) 分别根据不同的生物医学资源构建基因(蛋白质)、疾病、药物词典,利用基于特征耦合泛化的学习方法进行过滤。
(2) 分别根据不同的训练集,用词典加CRF的方法训练出标注模型。
(3) 对于给定生物医学文本,分别对基因(蛋白质)、疾病、药物实体进行识别。
(4) 把识别的结果进行合并,最后以可视化的形式给出对三种命名实体的识别结果。
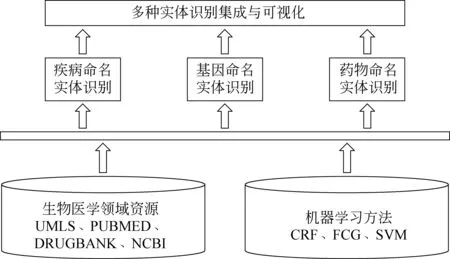
图1 系统框架图
2.2 基因(蛋白质)识别方法
基因(蛋白质)识别的处理过程主要分为三步: (1)基因(蛋白质)词典生成; (2)基因(蛋白质)词典过滤; (3)词典与CRF结合进行实体识别。
2.2.1 基因(蛋白质)词典生成
本文实验中基因(蛋白质)词典从ABGene lexicon和BioThesaurus 2.0两个生物医学相关的数据库中提取。我们发现得到的基因(蛋白质)词典条目很小,词典的覆盖面很低。这是由于基因(蛋白质)的词汇不仅数量巨大,而且包括很多变体名称,我们很难一一列举出同一基因(蛋白质)的所有表达形式。为了提高基因(蛋白质)词典的覆盖度,我们用简单的基于规则的方法生成了大量的基因(蛋白质)变体名称,将每个名字中的非数字和字母的符号去掉,并且分别去掉最左边或右边的词和二字串,将变换后的名字加入词典中,从而使词典得到了扩充。扩充词典虽然可以较大程度的提高词典覆盖度,却引入了大量的噪音,极大的影响了识别结果。因此我们用基于特征耦合泛化的学习方法[11]对词典进行去噪。
2.2.2 基因(蛋白质)词典过滤
在基因(蛋白质)词典去噪过程中,我们把词典去噪看作分类问题,采用的是由李彦鹏等提出的基于特征耦合泛化的学习方法,该方法可以有效地解决数据稀疏的问题。
该算法的基本思想是将特征共现的信息转化为新的特征。特征共现要涉及两种特征,一种是实例识别特征,一种是类别识别特征。根据这两种类型特征在未标注语料中的共现情况我们得到新的特征。相比于原始特征,新特征具有更丰富的信息量和更泛化的表示、更低的维度。而且这些新特征融合了未标注语料信息,弥补了有限训练集中信息量的不足。最后我们生成一个高覆盖度、低噪音的基因(蛋白质)词典。关于该算法的详细描述可参考文献[11]。
2.2.3 词典与CRF结合进行基因(蛋白质)命名实体识别
基于词典的方法简单实用,但受限于词典的规模和质量。CRF是一个无向图模型,用来标记和切分序列化数据的统计框架模型。由于CRF不需要遵循HMM严格的条件独立性,并且在一定程度上克服了MEMM的偏置问题,因此CRF更好地拟合了真实世界的数据,被广泛应用于自然语言处理任务中。而且作为机器学习方法的一种,CRF可以判别出生物实体数据库中未包含的实体,并且可以根据上下文环境对已登录词给出更准确的答案,比基于字典的方法更灵活。但CRF最大的一个缺陷在于,它依赖于训练集的规模和质量以及特征值的选取,识别性能仍有改进的空间。为了弥补单纯基于词典的方法的缺陷,并结合CRF方法的优势,本文提出了一种基于词典和CRF相结合的生物命名实体识别方法。实验结果表明,词典与CRF相结合方法的实体识别效果比仅用词典或仅用CRF方法效果要好。
2.3 疾病名识别方法
疾病名识别的处理过程主要分为三步:(1)词典生成;(2)词典与CRF结合进行实体识别;(3)用全称缩写对进行后处理。
2.3.1 疾病名词典生成
本文实验中疾病名词典从以下四个部分抽取:AZDC语料[12]、NCBI[13]训练集语料、药物基因组学*http://www.pharmgkb.org/downloads.jsp、以及UMLS的超级叙词表。由于疾病名词典相对基因(蛋白质)名比较规范,不需要去除噪音,因此我们分别直接从四个语料中抽取疾病名称,然后合并成一个词典,作为疾病名词典。词典与CRF结合对疾病命名实体进行识别与2.2.3节中方法相同,这里不再赘述。
2.3.2 用全称缩写对进行后处理
在疾病命名实体识别所用的生物文献中存在许多全称缩写词对,如“Ataxia-telangiectasia(A-T)”。经过统计,我们在训练集中发现共有396个全称缩写词对,其中267个被标注为疾病,占总数的67.42%。当疾病名实体第一次出现时一般用疾病名全称,以后会用其缩写词代替疾病名全称。如若只识别出疾病名全称而不对疾病名的缩写词进行识别,识别召回率会降低。为了充分利用疾病名全称缩写词对,我们使用全称缩写词对识别算法从测试集中提取与标注结果中的疾病名全称匹配的缩写词,然后再次扫描测试集把未识别出的缩写词识别出来。
表1给出了利用全称缩写词对进行调整的示例。我们采用IBO的标记模式对疾病名实体进行标注。标注为O代表疾病命名实体之外的词;I-XXX 标注是用于在疾病实体类型XXX之中的词;当两个类型为XXX的疾病实体直接相邻时,则后一个疾病实体的第一个词会被标注为B-XXX来表明这是另一个疾病实体的开始。
在表1中我们可以看出,在调整前“T-cell prolymphocytic leukaemia”被识别为一个疾病名,而它的缩写形式未被识别出来。由于两者存在全称缩写的关系,因此我们推断“T-PLL”也应该识别为一个疾病名。
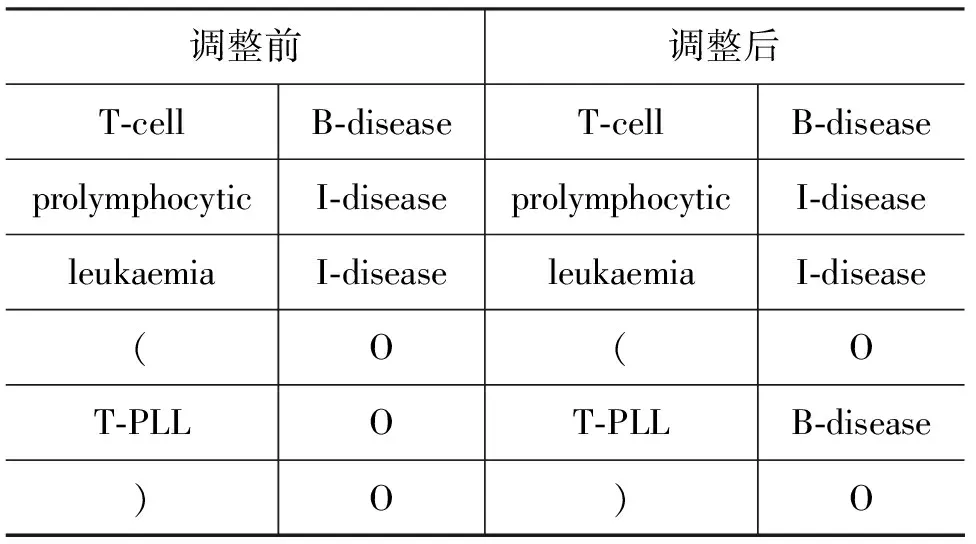
表1 利用全称缩写词对调整示例
2.4 药名识别方法
药名识别的处理过程主要分为三步:(1)药名词典生成;(2)药名词典过滤;(3)词典与CRF结合进行实体识别。
本文实验中药名词典种子来源于DRUGBANK。由于现有的药名种子较少,覆盖面较低,并且不能得到及时更新,因此为了提高覆盖面,我们根据药名种子生成了含有很多药名变体的药名词典,从而药名词典项得到了扩充。关于药名词典的具体构造过程可参考文献[14]。扩充词典虽然可以较大程度的提高词典覆盖度,却引入了大量的噪音,极大的影响了识别结果。因此,类似于基因(蛋白质)识别,我们也使用基于特征耦合泛化方法对词典进行去噪。之后用词典与CRF结合进行药物命名实体识别。其方法与2.2.2和2.2.3节中的描述的方法类似,这里不再赘述。
3 实验
3.1 基因(蛋白质)实体识别
在基因(蛋白质)词典生成过程中我们所用的数据库是ABGene lexicon和BioThesaurus 2.0两个生物医学相关的数据库。然后我们用简单的基于规则的方法生成了大量的基因(蛋白质)名变体名称,接着我们用基于特征耦合泛化的学习方法对词典进行过滤,最后生成一个高覆盖度、低噪音的基因(蛋白质)词典。
在基因(蛋白质)命名实体识别过程中,使用BioCreative II基因标准化任务的数据集,包括262篇测试摘要(其中含有785个人类基因标识符)和人类基因名同义词词典(32 975项)。关于该语料的详细介绍可参考文献[15]。
评测结果是基于标注结果和标准答案之间的匹配得到的。TP(True Positive)值指的是标注结果和标准答案可以匹配的基因(蛋白质)名数量,即正确的正例;FP(False Positive)值指的是标注结果中包含,但是在标准答案中匹配不到的基因(蛋白质)名数量,即错误正例;FN(False Negative)值指的是标准答案中没有被匹配的答案数,即错误负例。我们给出召回率(R)、精确度(P)和F值的计算方式:
(1)
(2)
(3)
在基因(蛋白质)命名实体识别实验中,我们使用BioCreative II的测试集作为测试集,以便与 Ando[16]等的方法进行比较。
表2给出了不同方法在BioCreative II测试集上识别的结果,其中CRF和词典相结合的方法(即表中“CRF+词典”项)是指将词典查找的结果作为特征加入到CRF中去。从表2中我们可以看出,词典匹配的方法优于CRF,这是因为我们通过机器学习的方法构建一个高质量的基因(蛋白质)名字典,而后直接通过字典匹配的方法识别文中的命名实体。从表2我们可以看出,词典与CRF结合的方法优于词典匹配和只用CRF的方法。这充分说明词典在基因(蛋白质)名实体识别中具有一定的辅助作用,而且词典和CRF具有互补效果。从表2我们可以看出,与Ando等方法相比,我们的词典与CRF相结合的方法得到的F值最高。这是因为我们不仅通过机器学习的方法构造了一个高质量的基因(蛋白质)词典,而且将词典查找的结果作为特征加入到CRF中去,使词典与CRF优势互补,识别效率更高。
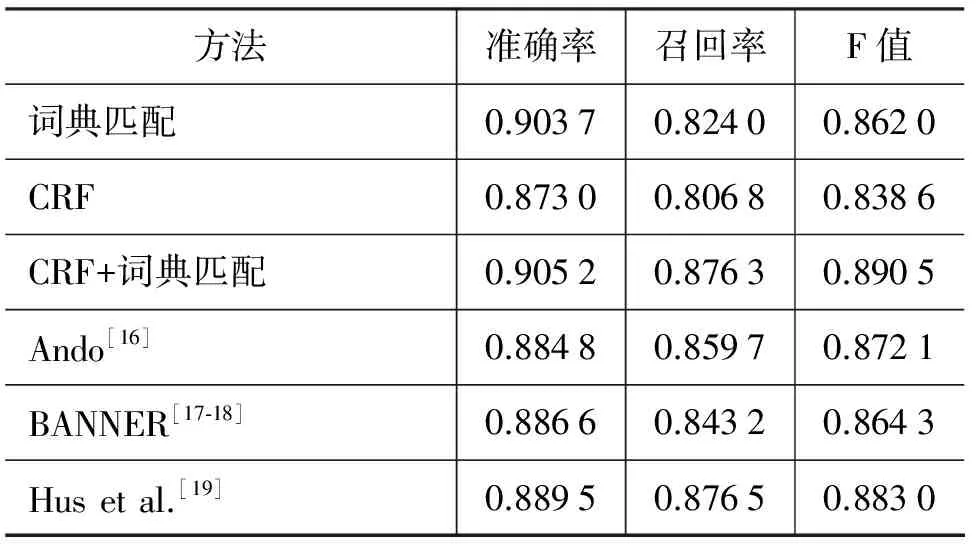
表2 BioCreative II测试集上的结果
3.2 疾病命名实体识别
在构建疾病名词典过程中,我们从AZDC语料[12]、NCBI[13]训练集语料、药物基因组学*http://www.pharmgkb.org/downloads.jsp里面抽取出疾病名,从而生成初步的疾病名词典。由于疾病名词典相对比较规范,不需要去除噪音,因此我们直接将抽取出来的词典作为疾病名词典。
在疾病名命名实体识别中,我们所采用的语料是NCBI。该语料来源于从793篇PUBMED摘要中提取的6 651个句子。其中,训练集中包括593篇摘要,测试集中包含100篇摘要,另外还有一个开发集,包含100篇摘要。

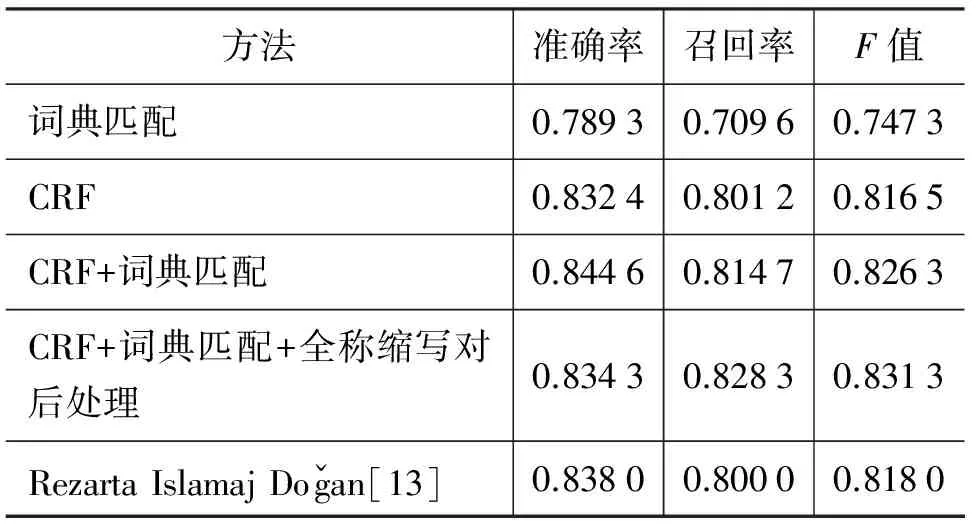
表3 NCBI测试集语料上的结果
3.3 药物命名实体识别
在构建药名词典过程中,我们首先从DRUGBANK中抽取药名作为种子,然后从PUBMED摘要中提取模板,用生成的模板再到PUBMED摘要中提取药名,从而生成初步的药名词典。最后用基于特征耦合泛化的学习方法对词典进行过滤,得到最终的药名词典。
在药物命名实体识别中,我们采用的训练集语料是DDIExtraction 2011评测的训练集语料*http://labda.inf.uc3m.es/DDIExtraction2011/dataset.html。语料是XML格式,每个句子中的药名已经识别出,所以我们可以将此语料作为训练集。我们用Segura-Bedmar等[20]用的语料*http://labda.inf.uc3m.es/DrugDDI/DrugNer.html作为测试集,以便与其方法进行比较。
表4给出了不同的方法在Segura-Bedmar等语料上识别的结果。从表4中我们可以看出,CRF优于词典匹配的方法。因为词典匹配使用的算法是前向最大匹配和后向最大匹配,所以识别出来的很多实体都只是部分匹配。另外,虽然我们用FCG的方法对药名词典进行去噪,但是词典中仍然存在一定的噪音没有消除,例如,“THIS”是一个药名的缩写,但是其小写形式“this”则只是英语中常用的代词。因此,词典匹配方法识别出的实体很多,但准确率和召回率并不高。从表4我们看出CRF和词典相结合的方法优于词典匹配和只用CRF方法,这充分说明词典在药名实体识别中具有一定的辅助作用,而且词典和CRF具有互补效果。与Segura-Bedmar等的Stem方法相比,不管是词典匹配、CRF还是词典匹配与CRF相结合的方法,结果都比Segura-Bedmar等的Stem方法要好。这是因为Stem方法在一定程度上限制了药名的范围,有些药名并不包含给定的词干,对于这样的药名Stem方法就无法识别出。另外有些词包含给定的词干但它并不是药名,这样的词相对于前者比较少。

表4 Segura-Bedmar语料上的结果
4 结论
本文介绍了一个面向生物医学领域的多实体识别系统MBNER。该系统可以在生物医学文本中同时识别出基因(蛋白质)、药物、疾病实体。其对基因(蛋白质)、药物、疾病实体的识别分别得到了89.05%,76.73%,90.12%的综合分类率(F-score)。图2给出了MBNER系统界面的示例图。该系统同时识别基因(蛋白质)、药物、疾病实体,并以不同的颜色对识别出的不同实体进行高亮显示。

图2 MBNER界面
命名实体识别是文本挖掘系统中的一个重要的基础步骤,在未来的工作中,我们将进一步提高生物医学文献中的命名实体识别准确程度,为其他文本挖掘技术奠定好的基础。
[1] 杨志豪. 面向生物医学领域的文本挖掘技术研究[D]. 大连:大连理工大学博士学位论文,2008.
[2] 王浩畅,李钰,赵铁军. 面向生物医学命名实体识别的多Agent元学习框架[J]. 计算机学报,2010,33(7):1256-1262.
[3] 彭春艳,张晖,包玲玉,等. 基于条件随机域的生物命名实体识别[J]. 计算机工程, 2009, 35(22): 197-199.
[4] 郑强,刘齐军,王正华,等. 生物医学命名实体识别的研究与进展[J]. 计算机应用研究,2010,27(3):811-815.
[5] Fukuda K, Tamura A, Tsunoda T, et al.Toward information extraction: Identifying protein names from
biological papers [J]. Pac Symp Biocomput, 1998: 707-718.
[6] Tuason O, Chen L, Liu H, et al.Biological nomenclatures: A source of lexical knowledge and ambiguity [J]. Pac Symp Biocomput, 2004, 9: 238-249.
[7] Tsochantaridis I, Hofmann T, Joachims T, et al.Support vector machine learning for interdependent and structured output spaces [C]//Proceedings of the twenty-first international conference on Machine learning. Banff, Alberta, Canada, July 04-08, 2004:104.
[8] Lin YF, Tsai TH, Chou WC, et al. A maximum entropy approach to biomedical named entity recognition [C]//Proceedings of the 4th ACM SIGKDD Workshop on Data Mining in Bioinformatics. Seattle, WA, 2004:56-61.
[9] Settles B. Biomedical named entity recognition using conditional random fields and rich feature sets [C]//Proceedings of the International Joint Workshop on Natural Language Processing in Biomedicine and Its Applications. Geneva, Switzerland, August 28-29, 2004 :104-107.
[10] Zhou GD, Su J. Named entity recognition using an HMM-based chunk tagger [C]//Proceedings of the 40th Annual Meeting on Association for Computational Linguistics, Philadelphia, Pennsylvania, July 07-12, 2002: 473-480.
[11] Li YP, Lin HF,Yang ZH. Incorporating rich background knowledge for gene named entity classification and recognition [J]. BMC Bioinformatics, 2009,10(223): 1-15.
[12] Md. Faisal Mahbub Chowdhury,Alberto Lavelli. Disease Mention Recognition with Specific Features[C]//Proceedings of the 2010 Workshop on Biome-dical Natural Language Processing, ACL 2010,Upp-sala, Sweden, 2010:83-90.

[14] 何林娜,杨志豪,林鸿飞,等. 基于特征耦合泛化的药名实体识别[C]//第八届全国信息检索学术会议论文集,2012.
[15] Wilbur J, Smith L and Tanabe L. BioCreative 2. gene mention task[C]//Proceedings of the Second BioCreative Challenge Evaluation Workshop, 2007, 7-16.
[16] Ando RK. BioCreative II gene mention tagging system at IBM Watson[C]//Proceedings of the Second BioCreative Challenge Evaluation Workshop,2007:101-103.
[17] Leaman R, Gonzalez G. BANNER: An executable survey of advances in biomedical named entity recognition[J]. Pac Symp Biocomput 2008, 13:652-663.
[18] Hakenberg J, Plake C, Leaman R,et al. Interspecies normalization of gene mentions with GNAT[J]. Bioinformatics 2008, 24:i126-132.
[19] Hsu CN, Chang YM, Kuo CJ, et al.Integrating high dimensional bi-directional parsing models for gene mention tagging[J]. Bioinformatics 2008, 24:i286-i294.
[20] Segura-Bedmar I, MartínezP, and Segura-Bedmar M. Drug name recognition and classification in biomedical texts: A case study outlining approaches underpinning automated systems [J]. Drug Discovery Today, 2008, 13(17-18): 816-823.
MBNER:Multiple Biomedical Named Entity Recognition System for Biomedical Literature
YANG Ya1, YANG Zhihao1, LIN Hongfei1, GONG Bendong2, WANG Jian1
(1. School of Computer Science and Technology, Dalian University of Technology, Dalian, Liaoning 116024, China;2. Dalian Branch of China National Tobacco Corporation, Dalian, Liaoning 116010, China)
Biological named entity recognition aims to find the name of the specified type in biomedical texts. At present, the research on entity recognition has been grown sharply because it is of particularly importance to extract biological entity information automatically from huge amounts of biomedical text. In this paper, we present a multiple entity recognition system in biomedical literature named MBNER (Multiple Biomedical Named Entity Recognizer), which can recognize gene (protein), drug and disease entities from biomedical texts simultaneously. This system can achieve an F-scores of 0.890 5, 0.767 3 and 0.901 2 on the gene (protein), drug and disease datasets, respectively. In addition, the system can visualize the recognition results of different named entities.
machine learning; feature coupling generalization; CRF; full abbreviation pairs

杨娅(1990—),硕士,主要研究领域为基于生物医学文献的实体识别。E⁃mail:yyang@mail.dlut.edu.cn杨志豪(1973—),教授,博士生导师,主要研究领域为文本挖掘、信息检索。E⁃mail:yangzh@dlut.edu.cn林鸿飞(1962—),教授,博士生导师,主要研究领域为搜索引擎、文本挖掘、情感计算和自然语言理解。E⁃mail:hflin@dlut.edu.cn
1003-0077(2016)01-0170-06
2013-07-08 定稿日期: 2014-05-16
国家自然科学基金(61070098, 61272373);中央高校基本科研业务费专项资金资助(DUT13JB09)
TP391
A
