RAISE技术改组转录因子RpoD调控大肠杆菌的低pH值耐受性
2016-04-25朱丽英梁世中
高 茜,朱丽英,周 伟,梁世中,江 凌,4
(1.南京工业大学生物与制药工程学院,江苏 南京 210009;2.南京工业大学化学与分子工程学院,江苏 南京 210009;
3.华南理工大学生物科学与工程学院,广东 广州 510641;4.南京工业大学食品与轻工学院,江苏 南京 210009)
RAISE技术改组转录因子RpoD调控大肠杆菌的低pH值耐受性
高茜1,朱丽英2,周伟1,梁世中3,江凌1,4
(1.南京工业大学生物与制药工程学院,江苏 南京 210009;2.南京工业大学化学与分子工程学院,江苏 南京 210009;
3.华南理工大学生物科学与工程学院,广东 广州 510641;4.南京工业大学食品与轻工学院,江苏 南京 210009)
摘要:在生物炼制生产过程中,微生物由于自身对酸的抗逆性差,其代谢生产过程会受到严重的影响。为解决该问题,本实验采用PCR方法从E.coli Dpα中扩增分离得到分子量为2 075 bp的RpoD完整序列(包含天然启动子区域和终止子区域),并利用RAISE方法,通过突变全局调控因子RpoD来提高大肠杆菌的耐酸能力。通过对104随机突变文库的多轮筛选,获得最佳突变菌株Mutant Ⅶ。在pH值为3.0、3.5、4.0、5.0、6.0 和7.0的条件下,对Mutant Ⅶ和对照菌株的生长情况进行比较分析,结果表明,在酸性条件下Mutant Ⅶ较对照菌株具有明显的生长优势,尤其在pH值为3.0时,相比对照菌株0.015 h(-1)的生长速率,Mutant Ⅶ的生长速率达到0.022 h(-1),具有较高应用价值。
关键词:大肠杆菌;耐酸性;RpoD;RAISE方法
生物炼制推动了可再生生物质资源转化为生物能源和化工产品的生物催化过程的发展。但是,由于现代生物炼制技术体系的生物质利用率低、产品定向合成能力差,严重制约了生物炼制生产过程的进一步发展。例如,谷氨酸生产中一般通过补加氨水控制pH值在7.0左右,待发酵结束后再加入浓硫酸使pH值降至3.2左右以利于有机酸提取。若将生产菌株的耐酸胁迫性从原始的pH值6.0提升至pH值4.6,则能显著减少中和剂的使用,降低下游污染。同时由于生产菌株酸耐受性的增强,最终产量会进一步提高。因此,提高菌种的耐酸能力有助于提高生物炼制生产过程的经济性和绿色指数[1]。
大肠杆菌(Escherichia coli)因具有细胞生长周期短、遗传背景清晰、培养操作简单等优点,已被用作丁二酸[2]、3-羟基丙酸[3]、乳酸[4]等有机酸生产的主要微生物。但是,在大规模有机酸发酵生产过程中,大肠杆菌的生长及其产品的产量都会受到酸根离子的影响[5]。因此,为了适应工业生产的需求,需进一步提高菌株的生长率。在大肠杆菌的不同生长环境中,其RNA聚合酶通过转录特异基因的选择性σ因子改变基因组的转录来调控整个生长过程。目前,大肠杆菌中发现的σ因子有7种,分别为:σD(σ70)、σN(σ54)、σS(σ38)、σH(σ32)、σF(σ28)、σE(σ24)和σfecI[6]。其中,σD是最主要的σ因子,负责与细胞生长相关的1 000多个基因的转录控制,是调控大肠杆菌耐酸性的最佳对象。
传统提高大肠杆菌耐酸性能的菌株构建方法(如物理/化学突变、适应性进化等)耗时耗力[7];而代谢工程突变方法,包括glpK、yqhD、gldA、pta、ldhA的敲除和cacKB、cscA、dhaB、dhaR、aldH的过表达都已经得到了广泛的应用[2-3]。但由于代谢途径的复杂性和多样性,在突变过程中很难获得一个全局最适表型。随机片段交换法(randominsertional/deletionalstrandexchangemutagenesis,RAISE)是一种新的突变文库构建方法,它汇集了多种DNA序列的突变形式,各种形式可以单独发生,也可以同时发生。同传统易错PCR构建突变文库的方法相比,不仅丰富了突变文库的突变形式,更提高了突变文库的筛选效率[8]。Fujii等[9]利用此法对TEM-1型β-内酰胺酶进行进化,经过3轮突变,使其对头孢他啶(ceftazidime)的最小抑制浓度提高了5 000倍。因此,以RAISE法替代易错PCR构建基因突变文库,不仅可以简化操作步骤,同时又能增强突变文库的多样性,大大提高工业微生物耐酸表型定向筛选的效率。
1实验
1.1材料与试剂
菌株E.coliDH5α,自行保存;质粒PMD18T-vector,TAKARA公司;pKSC[10],新加坡南阳理工大学蒋蓉蓉教授赠予。
酵母粉、蛋白胨,英国OXOID公司;DNAmarker、DNA凝胶回收试剂盒,TAKARA公司;卡那霉素,南京生兴生物有限公司;琼脂糖,Promega分装;其余试剂均为分析纯。
1.2方法
1.2.1总DNA的提取[11]
取1~4mLE.coliDH5α的过夜活化菌液,于12 000r·min-1离心2min,弃上清液。菌体沉淀物中加入150μL的TEbuffer(含RNaseA1),用移液枪吸打至菌体悬浮,提取总DNA。
1.2.2基因克隆和重组表达载体构建
根据GenBank公布的序列设计引物,下划线部分分别为EcoRI和HindⅢ的酶切位点:
上游引物F:CCGAATTCTGATTTAACGGCTTAAGTGCCGAAG。
下游引物R:TAAAGCTTGCCGGGTCGGCGGTAAC。
PCR条件为:94 ℃预变性5 min;94 ℃ 30 s,70 ℃ 30 s,72 ℃ 60 s,30个循环;72 ℃延伸5 min。
PCR扩增产物用DNA凝胶回收试剂盒回收目的片段,并与T载体连接,转化E.coliDpα,菌落PCR,挑取阳性克隆,提取质粒测序。
经测序无误后,小量抽提质粒,并用EcoRI和HindⅢ对质粒及表达载体pKSC进行双酶切,分别回收目的片段,连接转化E.coliDpα,挑取阳性克隆。
1.2.3突变文库的构建与筛选
采用RAISE方法构建突变文库。过夜活化菌株E.coliDpα-pKSC-rpoD,并小量抽提质粒,用EcoRI和HindⅢ对载体pKSC-rpoD进行双酶切。胶回收获得目的片段(2 075 bp),在含有Tris-HCl(pH=7.0)和MnCl2的缓冲体系中进行RNase-free DNase Ⅰ 酶切。为了避免在重叠PCR过程中产生过多的点突变,MnCl2作为DNase Ⅰ 的协同作用因子加入到整个体系中,以控制酶切文库的大小。根据琼脂糖凝胶电泳分析结果,在100~300 bp之间的酶切片段中加入EDTA终止酶切反应并进行胶回收。在含有胶回收产物、缓冲液和dNTPs的体系中,随机将TdT添加到目的片段的3′末端。由于DNA聚合酶没有校正能力,经胶回收获得的片段通过重叠PCR进行重新组装[8]。完整的RpoD突变片段(RAISE产物)进行PCR扩增。纯化后的产物用EcoRI和HindⅢ进行过夜酶切,连接、转化E.coliDpα。转化后的菌株涂布于含有30 μg·mL-1卡那霉素的LB平板上,并随机挑选白色克隆,转接入新鲜培养基中,即获得突变文库。
通过低pH值脉冲实验对液体突变文库进行表型筛选。过夜活化后的随机突变文库接种到50 mL的LB液体培养基中于37 ℃、200 r·min-1下培养。当菌株生长到对数生长期前期(OD600=0.3,下同)时,往培养基中加入36%的稀盐酸将pH值从4.0调至3.0,连续转接4次后,将菌株转接入新鲜培养基中,在37 ℃、200 r·min-1下培养2 h。用新鲜培养基冲洗菌株3次,并涂布于含有卡那霉素的LB平板上进行培养。待平板有单菌落长出时,随机挑选20个单菌落,提取质粒测序。为保证实验结果的准确性,经测序确定突变位点的RpoD突变片段与pKSC空质粒连接,转化E.coliDpα。具有最优耐酸表型的突变菌株将作为第一轮突变筛选的初始菌株,并转接入新鲜培养基中,培养基pH值分别从7.0降到3.0,重复3轮,以筛选出最优耐酸表型的突变菌株。具体方法见图1。
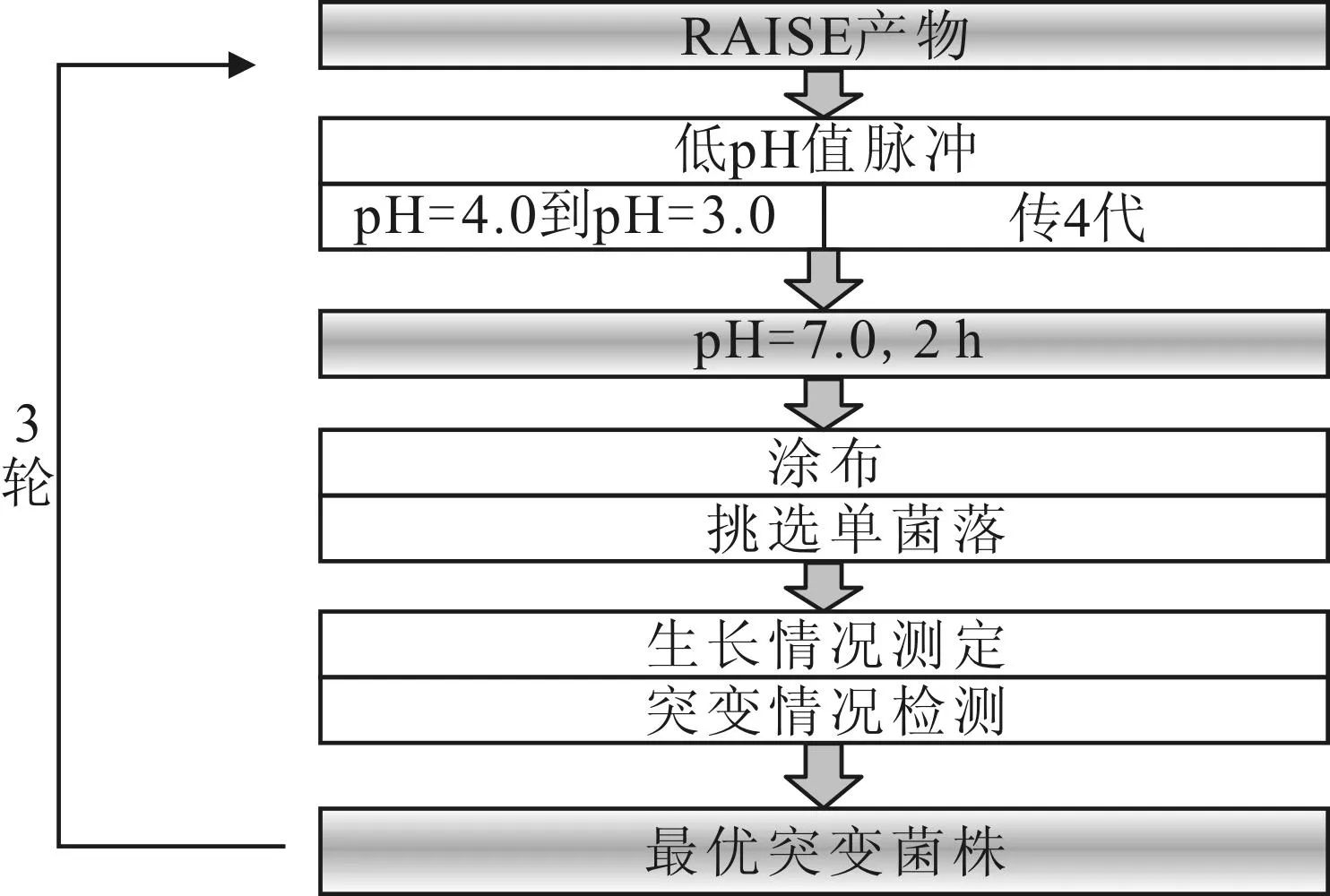
图1 突变文库的筛选方法
1.2.4耐酸性能的初步研究
过夜活化的突变菌株和对照菌株分别转接到LB新鲜培养基中,当菌株生长到对数生长期前期时,将培养基pH值分别调至3.0、3.5、4.0、5.0、6.0和7.0,用紫外分光光度计实时监测菌株在600 nm下的生长情况。
2结果与讨论
2.1RpoD表达载体的构建
首先,以E.coliDpα总DNA为模板,F、R为引物进行PCR扩增,得到分子量为2 075 bp的RpoD完整序列(包含天然启动子区域和终止子区域)。琼脂糖凝胶电泳显示,在2 000~3 000 bp之间有一条明显的条带,且无非特异性扩增(图2)。

M.5 000 bp DNA marker 1~10.平板上随机挑取的10处菌落
然后,将扩增所得目的片段回收,并与T载体连接,转化E.coliDpα。根据蓝白斑筛选方法随机挑取转化子,提取质粒测序。结果表明,得到的RpoD序列正确,和基因库中序列同源性100%、蛋白质同源性100%。
为了充分模拟自然条件下菌株的突变情况,本实验采用低拷贝质粒pKSC(拷贝数为1~5个,以pUC18质粒为骨架,复制子区域被pSC101的复制子替换,氨苄青霉素抗性被卡那霉素抗性替换)作为表达载体[5](图3)。利用EcoRI和HindⅢ对RpoD片段及表达载体pKSC进行酶切,琼脂糖凝胶电泳回收纯化。通过T4DNA连接酶将目的片段和表达载体连接,转化E.coliDpα,构建带有pKSC-rpoD重组载体的E.coliDpα-pKSC-rpoD初始工程菌株,即本实验的对照菌株(图4)。需要特别指出的是,由于pKSC质粒拷贝数偏低,并且大肠杆菌菌株基因组本身即含有rpoD基因,因此在PCR构建中容易产生假阳性菌株(对应图4的电泳槽1、2、7泳道)。
2.2突变文库的构建及筛选
以E.coliDpα-pKSC-rpoD为初始菌株,以低pH值为筛选指标,按1.2.3方法构建104的突变文库,重复3轮,分别以每轮获得的最优突变菌株为下一轮的初始菌株,并逐一进行突变文库筛选及突变菌株的分离纯化,测定每轮筛选出的突变菌株所能耐受的最低pH值,结果见图5。
由图5可看出,采用以上筛选方法,Mutant Ⅲ、Mutant Ⅵ和Mutant Ⅶ分别在第1、2和3轮突变文库筛选中耐酸性提升最明显,其中,Mutant Ⅶ在3轮突变文库构建及筛选过程中,因耐酸性能最优而被筛选为实验菌株,用于后续耐酸性能的进一步研究。

图3 pKSC质粒图谱

M.10 000 bp DNA marker 3~6.平板上随机挑取的4处单菌落

图5RpoD突变耐酸菌株的分离
Fig.5Isolation of acid tolerance RpoD factor mutants
2.3突变菌株突变位点的确定
根据以上突变文库的筛选结果,分别从3轮突变中获得Mutant Ⅲ、Mutant Ⅵ和Mutant Ⅶ3株耐酸性能提升显著的突变菌株,过夜活化,37 ℃、200 r·min-1下培养,提取质粒测序,以确定每轮突变中最优突变菌株的突变位点,结果见图6。
由图6可知,最优突变菌株的突变位点分别为:Mutant Ⅲ:P60S,D204G,S(603~607)deletion;Mutant Ⅵ:T95I,I287V,p18L,T(508~512)deletion;Mutant Ⅶ:F221L,I249F,I521V,E555V,D566N。

图6 每轮突变中最优突变菌株的突变位点
2.4突变菌株的耐酸性能
分别在pH值为3.0、3.5、4.0和7.0条件下,对实验菌株Mutant Ⅶ的倍增时间进行初步研究,并在pH值为3.0、3.5、4.0、5.0、6.0 和7.0条件下,对Mutant Ⅶ和对照菌株的耐酸性能进行进一步的比较分析,结果见图7、图8。
由图7可看出,菌株Mutant Ⅶ随着pH值的降低,倍增时间逐步增加,以pH=7.0时的倍增时间为0进行计算,当pH=3.0时,菌株的倍增时间达到27,但是,相比之前耐酸突变菌株的倍增时间[8],还有待进

图7 RAISE突变对每轮菌株耐酸性能的影响
一步缩短。由图7还可以看出,随着外部低pH值压
力的增加,菌株在酸性条件下的生长受到了明显抑制。
由图8可看出,在pH=7.0条件下,菌株Mutant Ⅶ与对照菌株的生长率没有明显差别,大约为0.900 h-1。同样的结果也出现在CRP的低pH值耐受性研究中[5];当菌株处于低pH值情况下时,突变菌株Mutant Ⅶ和对照菌株的生长都受到了明显的抑制,但Mutant Ⅶ的生长情况明显优于对照菌株:在pH=3.0时,Mutant Ⅶ的生长率为0.022 h-1,而对照菌株为0.015 h-1。当pH=4.0时,Mutant Ⅶ的生长率为0.034 h-1,明显高于之前研究中菌株的耐酸性能(0.012 h-1)[5]。表明,在低pH值条件下,Mutant Ⅶ比对照菌株具有非常明显的生长优势。此外,再次证明突变的σD因子能让菌株在低pH值条件下更稳定地生长。Mutant Ⅶ在所有突变菌株中耐酸性能最佳,但其耐酸机制仍有待进一步研究。

图8 Mutant Ⅶ和对照菌株在不同pH值条件下的生长情况
3结论
采用PCR方法,以E.coliDpα基因组DNA为模板,克隆出分子量为2 075 bp的σD编码基因RpoD,将其与表达载体pKSC连接,并转化E.coliDpα获得阳性重组子。经RAISE方法构建104的突变文库并以低pH值为压力进行3轮筛选,获得最优耐酸突变菌株Mutant Ⅶ。该菌株比对照菌株在低pH值条件下具有明显的生长优势,其优越的耐酸性能暗示了RAISE方法是一种能构建目的突变菌株的有效的突变方法,也进一步证明了突变后的全局转录调控因子RpoD能让菌株在低pH值压力条件下更稳定地生长。
参考文献:
[1]ELLIS D I,GOODACRE R.Metabolomics-assisted synthetic biology[J].Curr Opin Biotech,2012,23:22-28.
[2]CHAN S,KANCHANATAWEE S,JANTAMA K.Production of succinic acid from sucrose and sugarcane molasses by metabolically engineeredEscherichiacoli[J].Bioresour Technol,2012,103(1):329-336.
[3]KWAK S,PARK Y C,SEO J H.Biosynthesis of 3-hydroxypropionic acid from glycerol in recombinantEscherichiacoliexpressingLactobacillusbrevisdhaBanddhaR gene clusters andE.coliK-12aldH[J].Bioresour Technol,2013,135:432-439.
[4]MAZUMDAR S,BLANKSCHIEN M D,CLOMBURG J M,et al.Efficient synthesis of L-lactic acid from glycerol by metabolically engineeredEscherichiacoli[J].Microb Cell Fact,2013,12(1):7.
[5]CHUN A Y,LIANG Y X,ASHOK S,et al.Elucidation of toxicity of organic acids inhibiting growth ofEscherichiacoliW[J].Biotechnol Bioproc E,2014,19(5):858-865.
[6]ISHIHAMA A.Functional modulation ofEschrichiacoliRNA polymerase[J].Annu Rev Microbiol,2000,54:499-518.
[7]DRAGOSITS M,MATTANOVICH D.Adaptive laboratory evolution-principles and applications for biotechnology[J].Microb Cell Fact,2013,12:64.
[8]ALPER H,STEPHANOPOULOS G.Global transcription machinery engineering:A new approach for improving cellular phenotype[J].Metab Eng,2007,9(3):258-267.
[9]FUJII R,KITAOKA M,HAYASHI K.RAISE:A simple and novel method of generating random insertion and deletion mutations[J].Nucleic Acids Res,2006,34(4):e30.
[10]ZHANG H,CHONG H,CHING C B,et al.Random mutagenesis of global transcription factor cAMP receptor protein for improved osmotolerance[J].Biotech Bioeng,2012,109(5):1165-1172.
[11]WANG S X,WU X L,GONG W N,et al.Study on synthesis of trehalose by a novel microbial enzymatic system[J].Microbiology,2003,30(2):36-40.
Tailoring of Transcription Factor RpoD by RAISE to Regulate Low pH Value Tolerance ofEscherichiaColi
GAO Xi1,ZHU Li-ying2,ZHOU Wei1,LIANG Shi-zhong3,JIANG Ling1,4
(1.CollegeofBiotechnologyandPharmaceuticalEngineering,NanjingTechUniversity,Nanjing210009,China;2.CollegeofChemistryandMolecularEngineering,NanjingTechUniversity,Nanjing210009,China;3.SchoolofBioscienceandBioengineering,SouthChinaUniversityofTechnology,Guangzhou510641,China;4.CollegeofFoodScienceandLightIndustry,NanjingTechUniversity,Nanjing210009,China)
Abstract:In biorefinery processes,microbial metabolism was seriously suffered by the acid resistance of microbes themselves.To solve this problem,2 075 bp DNA fragment encoding RpoD,including promoter regions and termination regions,was cloned from E.coli DH5α by PCR method.And RAISE method was adopted to engineer the components of global regulator RpoD of E.coli to improve its acid tolerance.The best strain Mutant Ⅶ was identified from 104 random mutagenesis libraries based on the growth performance.Results showed that,under the pH value of 3.0,3.5,4.0,5.0,6.0 and 7.0 conditions,Mutant Ⅶ grew much better than the control strain.It was found that Mutant Ⅶ exhibited a significantly improved growth rate through multiwheel of acid-shock screening,which was 0.022 h(-1) as compared to that of 0.015 h(-1) with the control strain at pH value of 3.0.
Keywords:Escherichia coli;acid tolerance;RpoD;RAISE method
中图分类号:Q 754
文献标识码:A
文章编号:1672-5425(2016)03-0014-05
doi:10.3969/j.issn.1672-5425.2016.03.004
作者简介:高茜(1991-),女,江苏常州人,硕士研究生,研究方向:微生物学;通讯作者:江凌,博士,副教授,E-mail:jiangling@njtech.edu.cn。
收稿日期:2015-11-30
