Analysis of simple sequence repeats in rice bean (Vigna umbellata) using an SSR-enriched library
2016-04-05LixiWngKyungDoKimDongyingGoHonglinChenSuhuWngSukHLeeSottJksonXuzhenCheng
Lixi Wng, Kyung Do Kim, Dongying Go, Honglin Chen, Suhu Wng, SukH Lee, Sott A. Jkson, Xuzhen Cheng,*
aInstitute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing 100081, ChinabCenter for Applied Genetic Technologies, University of Georgia, 30602 Athens, GA, USAcPlant Genomics and Breeding Institute, Seoul National University, Seoul 151-921, Republic of Korea
Analysis of simple sequence repeats in rice bean (Vigna umbellata) using an SSR-enriched library
Lixia Wanga, Kyung Do Kimb, Dongying Gaob, Honglin Chena, Suhua Wanga, SukHa Leec, Scott A. Jacksonb, Xuzhen Chenga,*
aInstitute of Crop Science, Chinese Academy of Agricultural Sciences, Beijing 100081, China
bCenter for Applied Genetic Technologies, University of Georgia, 30602 Athens, GA, USA
cPlant Genomics and Breeding Institute, Seoul National University, Seoul 151-921, Republic of Korea
A R T I C L E I N F O
Article history:
Received 23 April 2015
Received in revised form
28 September 2015
Accepted 27 November 2015
Available online 4 December 2015
Keywords:
Vigna umbellata
SSR
Distribution and frequency
Mapping
Primer design
A B S T R A C T
Rice bean (Vigna umbellata Thunb.), a warm-season annual legume, is grown in Asia mainly for dried grain or fodder and plays an important role in human and animal nutrition because the grains are rich in protein and some essential fatty acids and minerals. With the aim of expediting the genetic improvement of rice bean, we initiated a project to develop genomic resources and tools for molecular breeding in this little-known but important crop. Here we report the construction of an SSR-enriched genomic library from DNA extracted from pooled young leaf tissues of 22 rice bean genotypes and developing SSR markers. In 433,562 reads generated by a Roche 454 GS-FLX sequencer, we identified 261,458 SSRs, of which 48.8% were of compound form. Dinucleotide repeats were predominant with an absolute proportion of 81.6%, followed by trinucleotides (17.8%). Other types together accounted for 0.6%. The motif AC/GT accounted for 77.7% of the total, followed by AAG/CTT (14.3%), and all others accounted for 12.0%. Among the flanking sequences, 2928 matched putative genes or gene models in the protein database of Arabidopsis thaliana, corresponding with 608 non-redundant Gene Ontology terms. Of these sequences, 11.2% were involved in cellular components, 24.2% were involved molecular functions, and 64.6% were associated with biological processes. Based on homolog analysis, 1595 flanking sequences were similar to mung bean and 500 to common bean genomic sequences. Comparative mapping was conducted using 350 sequences homologous to both mung bean and common bean sequences. Finally, a set of primer pairs were designed, and a validation test showed that 58 of 220 new primers can be used in rice bean and 53 can be transferred to mung bean. However, only 11 were polymorphic when tested on 32 rice bean varieties. We propose that this study lays the groundwork for developing novel SSR markers and will enhance the mapping of qualitative and quantitative traits and marker-assisted selection in rice bean and other Vigna species.
©2015 Crop Science Society of China and Institute of Crop Science, CAAS. Production and hosting by Elsevier B.V. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
* Corresponding author.
E-mail address: chengxuzhen@caas.cn (X. Cheng).
Peer review under responsibility of Crop Science Society of China and Institute of Crop Science, CAAS.
http://dx.doi.org/10.1016/j.cj.2015.09.004
2214-5141/©2015 Crop Science Society of China and Institute of Crop Science, CAAS. Production and hosting by Elsevier B.V. This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
1. Introduction
Rice bean (Vigna umbellata Thunb.) is a warm-season, annual, moderately short-lived legume. It is grouped into the large Vigna genus, which also includes other pulse crops such as adzuki bean (Vigna angularis Willd.), mung bean (Vigna radiata L.), andcowpea(Vignaunguiculata L.).Usedmainlyasadriedpulseor fodder, rice bean is often grown in hilly areas or on poor soil in East Asia [1]. In addition to its production of high-quality and nutritious grain [2–4], rice bean is highly tolerant to drought and acid soil [5,6] and is resistant to bruchid beetles [7] and several diseases [2]. These characteristics make this species an important genetic resource forcropbreedingin thefaceofthe frequent occurrence of climate extremes and increasing risk of pests and diseases. However, rice bean is a lesser known and underutilized legume, and most cultivars are landraces [8,9].
As members of one of the most economically and agriculturally important plant families, at least nine species of legume have had their whole genomes sequenced, including soybean [Glycine max (L.) Merr.] [10], common bean (Phaseolus vulgaris L.) [11], mung bean [12], and adzuki bean [13]. The latter two species are the most closely related to rice bean. Genome-wide synteny betweentheselegumeshasbeenreported[14–16]andrepresents a valuable resource for comparative analysis of legume genome evolution and the construction of high-resolution molecular linkagemapsforlegumeswhosegenomesarenotyetsequenced. Owing to the lack of molecular markers, genetic map construction [15,17] and diversity analysis [18,19] in rice bean have both been performed with markers transferred from related crops. No QTL for important traits have been reported to date.
Simple sequence repeats (SSRs), which are widely distributed in genomes [20,21], have been used extensively for genetic studies such as diversity evaluation [18,22,23], species identification [24,25], and gene mapping [26–28] because of their multi-allelic nature, reproducibility, co-dominant inheritance, and simplicity of detection. Increasing numbers of studies indicate that SSR-containing sequences play roles in gene function [29–31], a phenomenon that might prove helpful for mining gene-associated markers and accelerating the use of SSRs in marker-assisted selection for breeding. However, little information about SSRs in rice bean is known and no SSR markers have been developed specifically for rice bean.
With the development and decreased cost of highthroughput DNA sequencing, SSR markers have frequently been developed in many species with unavailable genome sequence by construction and sequencing of SSR-enriched libraries [32–38]. This approach has also provided an efficient way to improve understanding of SSRs in plant genomes. In this study, we constructed and sequenced an SSR-enriched library of rice bean and identified numerous SSRs in the sequencedata.Thenanalyzedthedistributionandfrequencyof SSRs, annotated the SSR-containing sequences, and mapped the flanking sequences to chromosomes of mung bean and common bean [11,12]. Finally, we designed a subset of new primers and validated their effectiveness and efficacy at amplifying polymorphic sequences. We report the first genome-wide SSR analysis and marker development for rice bean, and hope that will promote genetic studies and SSR marker-assisted breeding in this crop.
2. Materials and methods
2.1. Construction and sequencing of an SSR-enriched library
To identify variation in SSR sequences, fresh young leaves of 22 rice bean genotypes were collected and mixed (Table S1). Genomic DNA was extracted using the CTAB method [39]. The quality and quantity of DNA were assessed by agarose gel electrophoresis and a UV spectrophotometer, respectively.
An SSR-enriched library was constructed using methods derived from recent reports, with minor modifications [37,38,40,41]. Briefly, the genomic DNA was sheared mechanically by N2at 2.1 bar for 1.5 min, fragmented DNA between 500 and 800 bp in size was separated, polished with T4 DNA polymerase, and ligated to adaptors. Nested PCR was used to amplify the targeted fragments with primer sequences and then hybridized to oligo-probes consisting of SSR motifs. SSR-containing sequences adsorbed on streptavidin-coated magnetic beads were amplified by another round of PCR. Finally, products were isolated for sequencing with a Roche 454 GS-FLX instrument. The sequence data sets were processed to filter out weak and low-quality reads and to trim off the adaptor sequences using a series of normalization, correction and quality-filtering algorithms in the EMBOSS open software suite [42].
2.2. Identification of SSRs
SSRs were identified with the MISA (MIcroSAtellite) program with settings of 10, 6, 5, 5, 5, and 5 for minimum repeats of mono-, di-, tri-, tetra-, penta-, and hexanucleotides, respectively. Complementary sequences [(A)n= (T)n, (AC)n= (GT)n, and (ACC)n= (GGT)n] were included in calculating the ratios of repeat motifs. Two or more SSRs separated by no more than 100 bp were considered as a compound SSR.
2.3. Function prediction of SSR-containing sequences
As the genome sequence of rice bean is not yet available, we annotated the putative functions of the SSR-containing sequences using a homology-based method. SSRs were masked and only flanking sequences longer than 100 bp were selected to obtain more accurate prediction against the proteins of the annotated genes in Arabidopsis thaliana (http://www.phytozome. net/search.php) using BLASTx. All sequences with significant alignments (E-value≤10−4and similarity≥70%) were further considered and their putative functions were assigned to three independent Gene Ontology (GO) categories: biological process, cellular component, and molecular function, using BLAST2GO [43].
2.4. Mapping flanking sequences of rice bean SSR onto mung bean and common bean chromosomes
Of the legume crops that have had their whole genome sequenced, mung bean is the most closely related to rice bean, whereas the genome of common bean is the most intensively studied. To identify putative orthologous loci of rice bean SSRs in the two relatives, the flanking sequences (≥100 bp) of SSRswere searched forsequenceswith aspecifieddegreeofsimilarity in the databases of the reference genomes of mung bean and common bean, using BLASTn. Only sequences that met the following criteria were considered as putative loci of rice bean SSRs in both genomes: E-value≤1×10−10, sequence identity≥90%, and target sequence length≥80 bp. Genomic maps of mung bean and common bean were constructed with ArkMAP [44] based on the results of BLASTn.
2.5. Primer design and validation
Primers were designed based on SSR-containing sequences using Primer3 [45]. The major parameters were as follows: primer length was set as 17–27 bp, with 20 bp being optimal, PCR product size was set as 90–300 bp, optimum annealing temperature was set in the range 47–57°C, and the GC content of the primer was set as 30–65%, with 50% being optimal. The new primers were validated with a subset of primers using rice bean genomic DNA, and transferability to mung bean was assessed too. PCR was performed in a volume of 10 μL, containing 10 mmol L−1Tris–HCl, 50 mmol L−1KCl, 2 mmol L−1MgCl2, 100 mmol L−1of each dNTP, 0.4 mmol L−1of forward and reverse primer, 10 ng genomic DNA, and 0.5 U of Taq DNA polymerase. The reaction was performed with 35 cycles consisting of 30 s at 94°C for template denaturation, 30 s for primer annealing, and 30 s at 72°C for primer extension. Products were separated by 6% polyacrylamide gel electrophoresis using a DNA ladder to estimate fragment sizes, and visualized by silver staining [46]. Polymorphism of the markers that have clean and stable PCR fragments (here we call them effective primers) was further evaluated using 24 Chinese cultivated rice bean genotypes.
3. Results
3.1. Data set of an SSR-enriched library from the rice bean genome
A total of 433,562 reads were obtained, with a combined length of 102.05 Mb. We identified 261,458 SSRs, of which 48.8% (127,532) were in compound form. Only 119,523 SSRs were found to contain flanking sequences and to be applicable for primer design. In these SSRs, the frequencies of bases C, A, T, and G were 28.7%, 26.8%, 25.3%, and 19.2%, respectively.
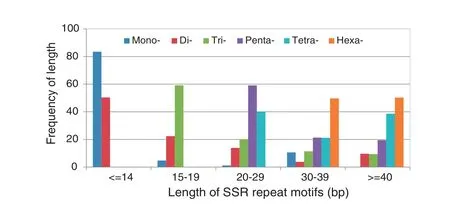
Fig. 1–Distribution of length of different SSR repeat types in rice genome based on a SSR-enriched library.
From the length distribution, it can be seen that the longer the sequence repeat motif, the lower was the frequency of mono-, di-, tri-, and pentanucleotides (Fig. 1). Most mononucleotide SSRs had fewer than 15 repeats (83.5%). For di-, tri-, and pentanucleotides,the majorityof SSRs(72.7%,79.3%,and80.5%, respectively) had fewer than 10 repeats. For tetranucleotide SSRs,61.4%wererepeated5–8times,andasimilarproportionof repeats lower (49.7%) or higher (50.3%) than six was observed in hexanucleotide SSRs.
3.2. Frequency and distribution of SSR in rice bean
Of the identified SSRs, dinucleotide repeats were the most frequent class (81.6%), followed by trinucleotide repeats (17.80%), while mono-, tetra-, penta-, and hexanucleotides accounted for only 0.10%, 0.13%, 0.05%, and 0.33%, respectively. Of mononucleotide motifs, 64.4% were A/T. AC/GT (95.2%) was the most abundant type of dinucleotide repeats, followed by AG/CT (4.6%). Of trinucleotide repeats, the most frequent type was AAG/CTT (80.2%), with AAC/GTT (13.6%) ranking second. Only three AAT/ATT and 11 CCG/CGG were observed. For tetra-, penta-, and hexanucleotide repeats, ACGT/ACGT (59.2%), AACAC/GTGTT (50.0%) and ACACGC/GCGTGT (21.8%), respectively, were more frequent than others (Fig. 2). Overall, AC/GT was the most common motif at (77.7%), 14.3% contained the AAG/CTT motif, and the remaining 8.0% contained other motifs.
3.3. Functional annotations of SSR-containing sequences
After the SSRs were masked, the flanking sequences (≥100 bp) were used to conduct BLASTx searches against the protein database of Arabidopsis. The flanking sequences of 2928 SSRs showed significant matches with gene models. These gene models were further found to correspond to 608 nonredundant GO terms. These GO terms can be classified into three groups: cellular components (11.2%), molecular functions (24.2%), and biological processes (64.6%). Sequences involved in cellular components were assigned to seven categories, with 64.7% being associated with“cytoplasmic components”. Eleven categories were assigned to molecular functions, with the majority (52.1%) classified as being involved in“enzyme activity”. Sequences associated with biological processes fell into 11 categories, of which“cellular processes”(25.7%) and “metabolic processes”(20.9%) were both prominent (Fig. 3).

Fig. 2–Distribution and frequency of different SSR motifs within each repeat type in the rice bean genome based on an SSR-enriched library.
3.4. Mapping of flanking sequences to mung bean and common bean genomes
The flanking sequences (≥100 bp) of rice bean SSRs were mapped to the mung bean and common bean genomes to determine the syntenic relationship between the three species. Of these flanking sequences, 1595 and 500 were mapped onto annotated chromosomes of mung bean and common bean. Another 572 and three flanking sequences were mapped onto scaffolds of mung bean and common bean, respectively (Fig. 4). Of the mapped flanking sequences, 350 were found in both species, showing the synteny between them (Fig. S1).
3.5. Primer design and validation
We designed a set of non-redundant primer pairs based on the flanking sequences of SSRs including 65.2% derived from trinucleotide repeats, 32.9% from dinucleotide repeats, and 1.9%fromotherrepeattypes.PCRvalidationshowedthat58ofa subset of 220 novel primers were effective in rice bean and that 53 could be successfully transferred to mung bean. Polymorphism tests showed that 11 yielded different alleles whentestedin32ricebeanaccessions,selectedatrandomfrom the Chinese collections. For example, we detected more than four distinct amplicons with the SSR marker Vum4-16 (Fig. 5). These multiple bands were likely derived from different alleles and suggested the genetic diversity of the rice bean germplasm.
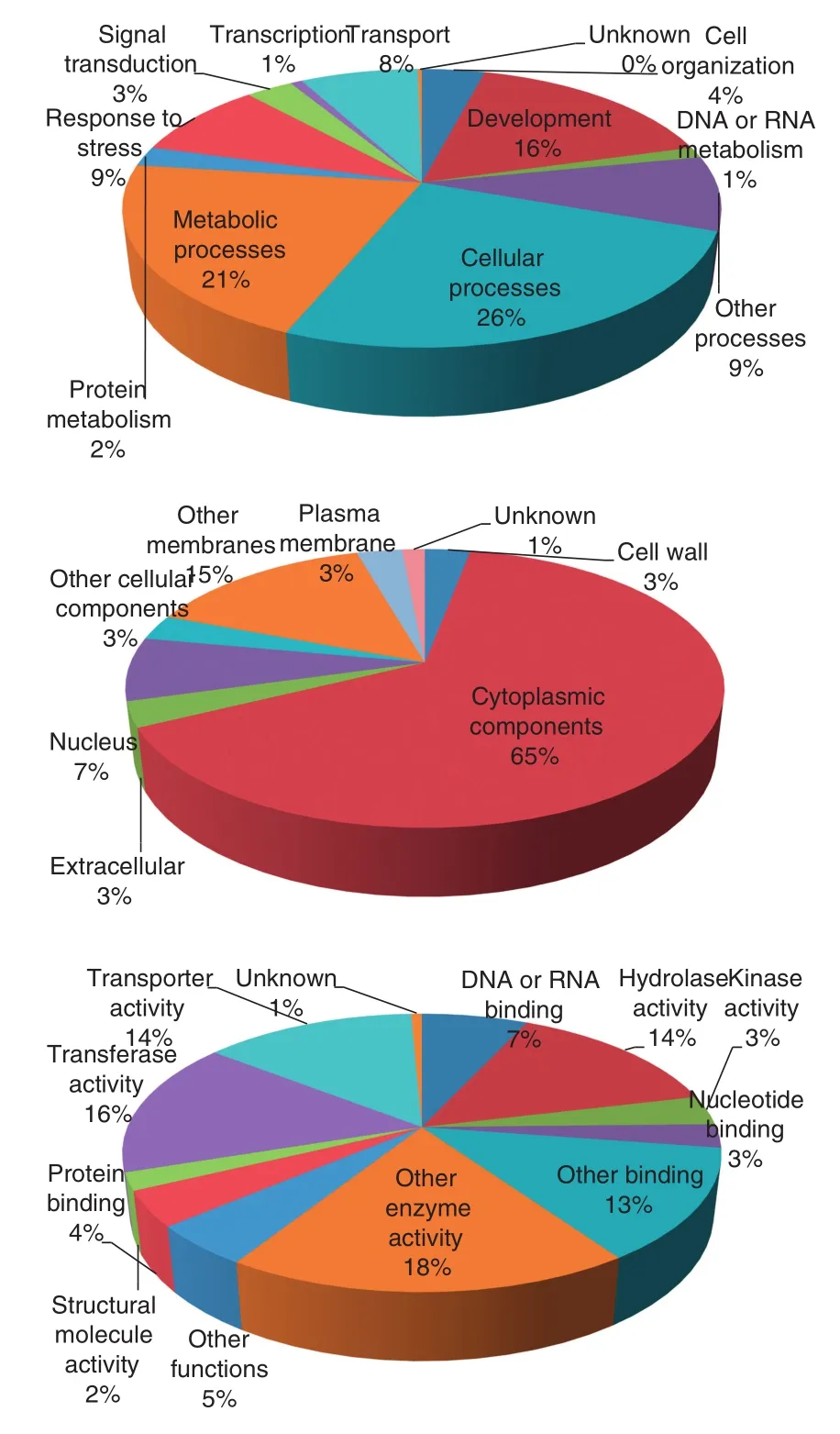
Fig. 3–Distribution of GO annotations of SSR flanking sequences from the rice bean genome.
4. Discussion
The frequency and distribution patterns of SSRs vary among different organisms. The predominant type in mushroom was mononucleotide [47], in Citrus trinucleotide [48], in cucumber tetranucleotide [49], and in cotton pentanucleotide [50]. The statistics of SSR distribution at genomic [37], transcriptional [51], and genic levels [52] in mung bean suggest that SSRs play different roles in inter- and intragenic sequences. Here we report the first study of SSR architecture in the rice bean genome. Dinucleotide repeatsaccountedfor thegreatestproportionof the total (81.6%), followed by trinucleotide repeats (17.8%). However, inthemungbeangenome,dinucleotidesandtrinucleotideseach accounted for 48.6% of total repeat types [37], although these two species both belong to subgenus Ceratotropis in genus Vigna.
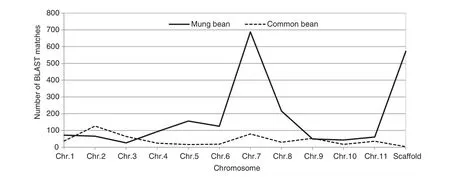
Fig. 4–Frequencies of nucleotide sequence matches of rice bean with genomes of mung bean and common bean by BLASTn alignment.
AC/TG was the most abundant (78.0%) motif in the rice bean genome, followed by AAG/TCC (14.0%). By contrast, in mung bean, AC/GT accounts for 41.8% and AAC/GTT accounts for 45.1% of all SSRs [37]. This difference can be explained by the higher proportion of dinucleotide (81.6%) than of trinucleotide (17.8%) repeats and higher percentage of AAG/CTT (80.2%) than AAC/GTT (13.6%) in trinucleotides in rice bean. The distribution of SSR motifs in rice bean also differs from that in faba bean [38]. However, for mononucleotide repeats, A/T, typical in most legumes [37,53] and other organisms [47,53], were also prevalent (64.4%) in rice bean. The frequency of motifs also varied when assessed indifferent data; for instance, AC/GT is the most prevalent dinucleotide motif in mung bean at the genomic level, whereas AG/CT dominates at the transcriptional level [51]. In addition, the motif frequencies in different region of genome also vary greatly [54,55], suggesting that different motifs play different roles in gene functions.
The gene annotation of SSR-containing sequences in rice bean suggested that many SSR-containing sequences were associated with two or more known category functions. The proportion of hits to non-redundant GO terms was much lower than that for EST (expressed sequence tags)-SSR reported in other species [48,56,57], and even lower than that in mung bean at the genome level [37]. Among these known GO annotations, more than half were associated with specific biological processes (64.6%) involving cellular and metabolic processes (46.6%), while only 11.2% and 24.2% were associated with cell components and molecular functions. In comparison with the results for mung bean, similar distributions of detailed annotations within the three categories were observed, possibly owing to the close relationship between the two crops.
To characterize the synteny between rice bean and related major crops, we mapped SSR flanking sequences to both the mung bean and common bean genomes. Owing to their relationships, different numbers of homologous sequences were identified between the two species, and the number of matches on chromosomes also varied greatly. From the distribution of sequences shared between mung bean and common bean, it can be seen that not all of the homologous sequences in common bean have matches in the mung bean genome. This finding may provide information about the evolution of the three species. A modest proportion (26.4%) of matches in mung bean genome matched with scaffolds, suggesting that the assembly of the mung bean reference genome needs to be improved. However, this comparative mapping will enhance the use of these sequences in developing molecular markers for genetic relationship and evolutionary research among species.
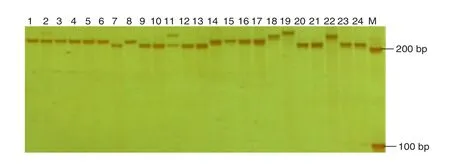
Fig. 5–Polymorphism of newly developed SSR primers using Chinese cultivated rice bean germplasm.
We designed a series of primer pairs based on SSR flanking sequences and validated a subset of primer pairs. The proportions of effectiveness and polymorphism were lower than had been observed in similar studies in legumes [37,38]. Although wecannotexplaintheloweffectiveness,thelowpolymorphism maybe due to the low diversity of samples used, given that the32 germplasm accessions were all collected in China. A study of SSR markers in Chinese mung bean similarly showed limited diversity[19].Infact,lowdiversityin Vignagermplasmhasbeen reported for several species, including both mung bean [58–63] and adzuki bean [58–63], except when the tested samples were selected from a wide geographical region [58–63] or wild genotypes were included [18].
The low polymorphism of Chinese rice bean collections also indicates that it is imperative to broaden the genetic pool by plant introduction to provide more elite germplasm for rice bean breeding. In addition, increasing the polymorphism of markers by including wild genotypes in diversity analyses or crosses in population construction is recommended, as these measures have proven to be effective in several studies [15,64–66].Inthepresentstudy,thesetofnovelprimerswehave described can be transferred to mung bean and will contribute to comparative mapping between these two species.
Supplementary material to this article can be found online at http://dx.doi.org/10.1016/j.cj.2015.09.004.
Acknowledgments
This study was supported by earmarked funds for China Agriculture Research System (CARS-09) and the Agricultural Science and Technology Innovation Program (ASTIP).
R E F E R E N C E S
[1] N. Tomooka, The origin of rice bean (Vigna umbellata) and azuki bean (V. angularis): the evolution of two lesser-known Asian beans, in: T. Akimichi (Ed.), An Illustrated Eco-history of the Mekong River Basin, White Lotus Co. Ltd., Bangkok, Thailand. 2009, pp. 33–35.
[2] K.P. Chandel, B.S. Joshi, R.K. Arora, K.C. Part, Rice bean–a new pulse with high potential, Indian Farm. 28 (1978) 19–22.
[3] R. Katoch, Nutritional potential of rice bean (Vigna umbellata): an underutilized legume, J. Food Sci. 78 (2013) C8–C16.
[4] Y. Yao, X.Z. Cheng, L.X. Wang, S.H. Wang, G.X. Ren, Major phenolic compounds, antioxidant capacity and antidiabetic potential of rice bean (Vigna umbellata L.), China, Intl. J. Mol. Sci., 13 2012, pp. 2707–2716.
[5] G.K. Dwivedi, Tolerance of some crops to soil acidity and response to liming, J. Indian Soc. Soil Sci. 44 (1996) 736–743.
[6] W. Fan, H.Q. Lou, Y.L. Gong, M.Y. Liu, Z.Q. Wang, J.L. Yang, S.J. Zheng, Identification of early Al-responsive genes in rice bean (Vigna umbellata) roots provides new clues to molecular mechanisms of Al toxicity and tolerance, Plant Cell Environ. 37 (2014) 1586–1597.
[7] K. Kashiwaba, N. Tomooka, A. Kaga, O.K. Han, D.A. Vaughan, Characterization of resistance to three bruchid species (Callosobruchus spp. Coleoptera, Bruchidae) in cultivated rice bean (Vigna umbellata), J. Econ. Entomol. 96 (2003) 207–213.
[8] B.L. Thaware, V.W. Bendale, V.A. Toro, Konkan rice bean-1 (Rb-10), a new fodder rice bean variety for Konkan region of Maharashtra, J. Maharashtra Agric. Univ. 30 (2005) 295–298.
[9] X.Z. Cheng, S.M. Wang, The History of Chinese Food Legume Varieties, China Agricultural Science and Technology Press, Beijing, 2009 (in Chinese).
[10] J. Schmutz, S.B. Cannon, J. Schlueter, J.X. Ma, T. Mitros, W. Nelson, D.L. Hyten, Q.J. Song, J.J. Thelen, J.L. Cheng, D. Xu, U. Hellsten, G.D. May, Y. Yu, T. Sakurai, T. Umezawa, M.K. Bhattacharyya, D. Sandhu, B. Valliyodan, E. Lindquist, M. Peto, D. Grant, S.Q. Shu, D. Goodstein, K. Barry, M. Futrell-Griggs, B. Abernathy, J.C. Du, Z.X. Tian, L.C. Zhu, N. Gill, T. Joshi, M. Libault, A. Sethuraman, X.C. Zhang, K. Shinozaki, H.T. Nguyen, R.A. Wing, P. Cregan, J. Specht, J. Grimwood, D. Rokhsar, G. Stacey, R.C. Shoemaker, S.A. Jackson, Genome sequence of the palaeopolyploid soybean, Nature 463 (2010) 178–183.
[11] J. Schmutz, P.E. McClean, S. Mamidi, G.A. Wu, S.B. Cannon, J. Grimwood, J. Jenkins, S.Q. Shu, Q.J. Song, C. Chavarro, M. Torres-Torres, V. Geffroy, S.M. Moghaddam, D.Y. Gao, B. Abernathy, K. Barry, M. Blair, M.A. Brick, M. Chovatia, P. Gepts, D.M. Goodstein, M. Gonzales, U. Hellsten, D.L. Hyten, G.F. Jia, J.D. Kelly, D. Kudrna, R. Lee, M.M.S. Richard, P.N. Miklas, J.M. Osorno, J. Rodrigues, V. Thareau, C.A. Urrea, M. Wang, Y. Yu, M. Zhang, R.A. Wing, P.B. Cregan, D.S. Rokhsar, S.A. Jackson, A reference genome for common bean and genome-wide analysis of dual domestications, Nat. Genet. 46 (2014) 707–713.
[12] Y.J. Kang, S.K. Kim, M.Y. Kim, P. Lestari, K.H. Kim, B.K. Ha, T.H. Jun, W.J. Hwang, T. Lee, J. Lee, S. Shim, M.Y. Yoon, Y.E. Jang, K.S. Han, P. Taeprayoon, N. Yoon, P. Somta, P. Tanya, K.S. Kim, J.G. Gwag, J.K. Moon, Y.H. Lee, B.S. Park, A. Bombarely, J.J. Doyle, S.A. Jackson, R. Schafleitner, P. Srinives, R.K. Varshney, S.H. Lee, Genome sequence of mungbean and insights into evolution within Vigna species, Nat. Commun. 5443 (2014).
[13] Y.J. Kang, D. Satyawan, S. Shim, T. Lee, J. Lee, W.J. Hwang, S.K. Kim, P. Lestari, K. Laosatit, K.H. Kim, T.J. Ha, A. Chitikineni, M.Y. Kim, J.M. Ko, J.G. Gwag, J.K. Moon, Y.H. Lee, B.S. Park, R.K. Varshney, S.H. Lee, Draft genome sequence of adzuki bean, Vigna angularis, Sci. Rep. 5 (2015) 8069.
[14] D. Zhao, X.Z. Cheng, L.X. Wang, S.H. Wang, Y.L. Ma, Integration of mungbean (Vigna radiata) genetic linkage map, Acta Agron. Sin. 36 (2010) 932–939 (in Chinese with English abstract).
[15] T. Isemura, A. Kaga, N. Tomooka, T. Shimizu, D.A. Vaughan, The genetics of domestication of rice bean, Vigna umbellata, Ann. Bot. 106 (2010) 927–944.
[16] P.E. McClean, S. Mamidi, M. McConnell, S. Chikara, R. Lee, Synteny mapping between common bean and soybean reveals extensive blocks of shared loci, BMC Genomics 11 (2010).
[17] P. Somta, A. Kaga, N. Tomooka, K. Kashiwaba, T. Isemura, B. Chaitieng, P. Srinives, D.A. Vaughan, Development of an interspecific Vigna linkage map between Vigna umbellata (Thunb.) Ohwi & Ohashi and V-nakashimae (Ohwi) Ohwi & Ohashi and its use in analysis of bruchid resistance and comparative genomics, Plant Breed. 125 (2006) 77–84.
[18] J. Tian, T. Isemura, A. Kaga, D.A. Vaughan, N. Tomooka, Genetic diversity of the rice bean (Vigna umbellata) genepool as assessed by SSR markers, Genome 56 (2013) 717–727.
[19] L.X. Wang, H.L. Chen, P. Bai, J.X. Wu, S.H. Wang, M.W. Blair, X.Z. Cheng, The transferability and polymorphism of mung bean SSR markers in rice bean germplasm, Mol. Breed. 35 (2015) 77.
[20] M. Morgante, A.M. Olivieri, PCR-amplified microsatellites as markers in plant genetics, Plant J. 3 (1993) 175–182.
[21] G. Tóth, Z. Gáspári, J. Jurka, Microsatellites in different eukaryotic genomes: survey and analysis, Genome Res. 10 (2000) 967–981.
[22] B.W. Legesse, A.A. Myburg, K.V. Pixley, A.M. Botha, Genetic diversity of African maize inbred lines revealed by SSR markers, Hereditas 144 (2007) 10–17.
[23] L.X. Wang, R.X. Guan, Z.X. Liu, R.Z. Chang, L.J. Qiu, Genetic diversity of Chinese cultivated soybean revealed by SSR markers, Crop Sci. 46 (2006) 1032–1038.
[24] D.J. Perry, Identification of Canadian durum wheat varieties using a single PCR, Theor. Appl. Genet. 109 (2004) 55–61.
[25] F. Martin, An application of kernel methods to variety identification based on SSR markers genetic fingerprinting, BMC Bioinf. 12 (2011) 177.
[26] S.K. Biradar, R.M. Sundaram, T. Thirumurugan, J.S. Bentur, S. Amudhan, V.V. Shenoy, B. Mishra, J. Bennett, N.P. Sarma, Identification of flanking SSR markers for a major rice gall midge resistance gene Gm1 and their validation, Theor. Appl. Genet. 109 (2004) 1468–4173.
[27] Y.H. Wang, D.D. Poudel, K.H. Hasenstein, Identification of SSR markers associated with saccharification yield using pool-based genome-wide association mapping in sorghum, Genome 54 (2011) 883–889.
[28] O.A. Gutierrez, A.F. Robinson, J.N. Jenkins, J.C. McCarty, M.J. Wubben, F.E. Callahan, R.L. Nichols, Identification of QTL regions and SSR markers associated with resistance to reniform nematode in Gossypium barbadense L. accession GB713, Theor. Appl. Genet. 122 (2011) 271–280.
[29] Y.C. Li, A.B. Korol, T. Fahima, A. Beiles, E. Nevo, Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review, Mol. Ecol. 11 (2002) 2453–2465.
[30] T.W. Hefferon, J.D. Groman, C.E. Yurk, G.R. Cutting, A variable dinucleotide repeat in the CFTR gene contributes to phenotype diversity by forming RNA secondary structures that alter splicing, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 3504–3509.
[31] C. Schlotterer, Genome evolution: are microsatellites really simple sequences? Curr. Biol. 8 (1998) R132–R134.
[32] J. Li, M. Båga, P. Hucl, R. Chibbar, Development of microsatellite markers in canary seed (Phalaris canariensis L.), Mol. Breed. 28 (2010) 611–621.
[33] J.M. Soriano, E. Zuriaga, P. Rubio, G. Llácer, R. Infante, M.L. Badenes, Development and characterization of microsatellite markers in pomegranate (Punica granatum L.), Mol. Breed. 27 (2011) 119–128.
[34] M. Das, S. Banerjee, R. Dhariwal, S. Vyas, R.R. Mir, N. Topdar, A. Kundu, J.P. Khurana, A.K. Tyagi, D. Sarkar, M.K. Sinha, H.S. Balyan, P.K. Gupta, Development of SSR markers and construction of a linkage map in jute, J. Genet. 91 (2012) 21–31.
[35] C. Tan, Y. Wu, C.M. Taliaferro, G.E. Bell, D.L. Martin, M.W. Smith, Development and characterization of genomic SSR markers in Cynodon transvaalensis Burtt-Davy, Mol. Genet. Genomics 289 (2014) 523–531.
[36] C.D. Ishibashi, A.R. Shaver, D.P. Perrault, S.D. Davis, R.L. Honeycutt, Isolation of microsatellite markers in a chaparral species endemic to southern California, Ceanothus megacarpus (Rhamnaceae), Appl. Plant Sci. 1 (5) (2013) 1200393.
[37] L.X. Wang, M. El baidouri, B. Abernathy, H.L. Chen, S.H. Wang, S.H. Lee, S.A. Jackson, X.Z. Cheng, Distribution and analysis of SSR in mung bean (Vigna radiata L.) genome based on an SSR-enriched library, Mol. Breed. 35 (2015) 25.
[38] T. Yang, S. Bao, R. Ford, T. Jia, J. Guan, Y. He, X. Sun, J. Jiang, J. Hao, X. Zhang, X. Zong, High-throughput novel microsatellite marker of faba bean via next generation sequencing, BMC Genomics 13 (2012) 602.
[39] J.J. Doyle, J.L. Doyle, A rapid DNA isolation procedure for small quantities of fresh leaf tissue, Phytochem. Bull. 19 (1987) 11–15.
[40] T.C. Glenn, N.A. Schable, Isolating microsatellite DNA loci, Methods Enzymol. 395 (2005) 202–222.
[41] Z. Gao, J. Wu, Z. Liu, L. Wang, H. Ren, Q. Shu, Rapid microsatellite developmentfortreepeony and itsimplications, BMC Genomics 14 (2013) 886.
[42] P. Rice, I. Longden, A. Bleasby, EMBOSS: the European molecular biology open software suite, Trends Genet. 16 (2000) 276–277.
[43] A. Conesa, S. Gotz, J.M. Garcia-Gomez, J. Terol, M. Talon, M. Robles, Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research, Bioinformatics 21 (2005) 3674–3676.
[44] T. Trevor, A. Lawn, ArkMAP: integrating genomic maps across species and data sources, BMC Bioinf. 14 (2013) 246.
[45] S. Rozen, H. Skaletsky, Primer3 on the WWW for general users and for biologist programmers, Methods Mol. Biol. 132 (2000) 365–386.
[46] A. Ayana, E. Bekele, T. Bryngelsson, Genetic variation in wild sorghum (Sorghum bicolor ssp. verticilliflorum (L.) Moench) germplasm from Ethiopia assessed by random amplified polymorphic DNA (RAPD), Hereditas 132 (2000) 249–254.
[47] H. Sonah, R.K. Deshmukh, A. Sharma, V.P. Singh, D.K. Gupta, R.N. Gacche, J.C. Rana, N.K. Singh, T.R. Sharma, Genome-wide distribution and organization of microsatellites in plants: an insight into marker development in Brachypodium, PLoS One 6 (2011), e21298.
[48] S.R. Liu, W.Y. Li, D. Long, C.G. Hu, J.Z. Zhang, Development and characterization of genomic and expressed SSRs in Citrus by genome-wide analysis, PLoS One 8 (2013).
[49] P.F. Cavagnaro, D.A. Senalik, L.M. Yang, P.W. Simon, T.T. Harkins, C.D. Kodira, S.W. Huang, Y.Q. Weng, Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.), BMC Genomics 11 (2010) 569.
[50] C. Zou, C. Lu, Y. Zhang, G. Song, Distribution and characterization of simple sequence repeats in Gossypium raimondii genome, Bioinformation 8 (2012) 801–806.
[51] K.T. Moe, J.W. Chung, Y.I. Cho, J.K. Moon, J.H. Ku, J.K. Jung, J. Lee, Y.J. Park, Sequence information on simple sequence repeats and single nucleotide polymorphisms through transcriptome analysis of mungbean, J. Integr. Plant Biol. 53 (2011) 63–73.
[52] S.K. Gupta, R. Bansal, T. Gopalakrishna, Development and characterization of genic SSR markers for mungbean (Vigna radiata (L.) Wilczek), Euphytica 195 (2014) 245–258.
[53] M.W. Blair, N. Hurtado, C.M. Chavarro, M.C. Munoz-Torres, M.C. Giraldo, F. Pedraza, J. Tomkins, R. Wing, Gene-based SSR markers for common bean (Phaseolus vulgaris L.) derived from root and leaf tissue ESTs: an integration of the BMc series, BMC Plant Biol. 11 (2011) 50.
[54] J.H. Mun, D.J. Kim, H.K. Choi, J. Gish, F. Debelle, J. Mudge, R. Denny, G. Endre, O. Saurat, A.M. Dudez, G.B. Kiss, B. Roe, N.D. Young, D.R. Cook, Distribution of microsatellites in the genome of Medicago truncatula: a resource of genetic markers that integrate genetic and physical maps, Genetics 172 (2006) 2541–2555.
[55] J. Qian, H. Xu, J. Song, J. Xu, Y. Zhu, S. Chen, Genome-wide analysis of simple sequence repeats in the model medicinal mushroom Ganoderma lucidum, Gene 512 (2013) 331–336.
[56] W. Guo, C. Cai, C. Wang, Z. Han, X. Song, K. Wang, X. Niu, K. Lu, B. Shi, T. Zhang, A microsatellite-based, gene-rich linkage map reveals genome structure, function and evolution in Gossypium, Genetics 176 (2007) 527–541.
[57] D. Shen, H. Sun, M. Huang, Y. Zheng, Y. Qiu, X. Li, Z. Fei, Comprehensive analysis of expressed sequence tags from cultivated and wild radish (Raphanus spp.), BMC Genomics 14 (2013) 721.
[58] L.X. Wang, X.Z. Cheng, S.H. Wang, H. Liang, D. Zhao, N. Xu, Genetic diversity of adzuki bean germplasm resources revealed by SSR markers, Acta Agron. Sin. 35 (2009) 1858–1865 (in Chinese with English abstract).
[59] M. Mimura, K. Yasua, H. Yamaguchi, RAPD variation in wild, weedy and cultivated adzuki beans in Asia, Genet. Resour. Crop. Evol. 47 (2000) 603–610.
[60] A. Kaga, K. Hosaka, T. Kimura, S. Misoo, O. Kamijima, Application of random amplified polymorphic DNA (RAPD) analysis for adzuki bean and its related genera, Sci. Rep. Fac. Agric. 20 (1993) 171–176.
[61] H.X. Xu, T. Jing, N. Tomooka, A. Kaga, T. Isemura, D.A. Vaughan, Genetic diversity of the azuki bean (Vigna angularis (Willd.) Ohwi & Ohashi) gene pool as assessed by SSR markers, Genome 51 (2008) 728–738.
[62] Y. Liu, X.Z. Cheng, L.X. Wang, S.H. Wang, P. Bai, C.S. Wu, Genetic diversity research of mungbean germplasm resources by SSR markers in China, Sci. Agric. Sin. 46 (2013) 4197–4209 (in Chinese with English abstract).
[63] C. Sangiri, A. Kaga, N. Tomooka, D. Vaughan, P. Srinives, Genetic diversity of the mungbean (Vigna radiata, Leguminosae) genepool on the basis of microsatellite analysis, Aust. J. Bot. 55 (2007) 837–847.
[64] B. Chaitieng, A. Kaga, N. Tomooka, T. Isemura, Y. Kuroda, D.A. Vaughan, Development of a black gram [Vigna mungo (L.) Hepper] linkage map and its comparison with an azuki bean [Vigna angularis (Willd.) Ohwi and Ohashi] linkage map, Theor. Appl. Genet. 113 (2006) 1261–1269.
[65] O.K. Han, A. Kaga, T. Isemura, X.W. Wang, N. Tomooka, D.A. Vaughan, A genetic linkage map for azuki bean [Vigna angularis (Willd.) Ohwi & Ohashi], Theor. Appl. Genet. 111 (2005) 1278–1287.
[66] M.E. Humphry, C.J. Lambrides, S.C. Chapman, E.A.B. Aitken, B.C. Imrie, R.J. Lawn, C.L. McIntyre, C.J. Liu, Relationships between hard-seededness and seed weight in mungbean (Vigna radiata) assessed by QTL analysis, Plant Breed. 124 (2005) 292–298.
杂志排行

The Crop Journal的其它文章
- 7th International Crop Science Congress Announcement
- Editorial Board of The Crop Journal
- Comparisons of phaseolin type and α-amylase inhibitor in common bean (Phaseolus vulgaris L.) in China
- Genotypic variation for seed protein and mineral content among post-rainy season-grown sorghum genotypes
- Fosmid library construction and screening for the maize mutant gene Vestigial glume 1
- Intra-population genetic variance for grain iron and zinc contents and agronomic traits in pearl millet