Method for Control of Particle Size and Morphology of Paraf fi n/Polystyrene-Divinylbenzene Microcapsules
2016-03-22
(School of Chemistry, Beijing Institute of Technology, Beijing 100081)
Method for Control of Particle Size and Morphology of Paraf fi n/Polystyrene-Divinylbenzene Microcapsules
Wang Lili; Zhao Tianbo; Li Yang; Ding Hongjing
(School of Chemistry, Beijing Institute of Technology, Beijing 100081)
Microencapsulation of phase change materials (MicroPCMs) has been paid special attention because of their extensive applications in saving and releasing energy. MicroPCMs containing paraf fi n with a melting point of 55 ℃ in polystyrene-divinylbenzene (P(St-DVB)) were prepared by suspension-like polymerization. The characterization of microcapsules by FTIR, DSC and TG proved that paraf fi n had been successfully encapsulated and the proportion of encapsulated paraf fi n was 49.8%—58.5%. The effects of polyvinylpyrrolidone (PVP) with different molecular weights serving as the suspension stabilizer were investigated in detail. The results illustrated that the type of PVP had a signi fi cant in fl uence on the particle size of MicroPCMs. The average diameter of MicroPCMs decreased with an increasing molecular weight of PVP. Moreover, the crosslinker-postaddition method was adopted in this study to improve the morphology of P(St-DVB) MicroPCMs. SEM images showed that when the DVB was added at the 2nd hour of polymerization the morphology of obtained P(St-DVB) MicroPCMs exhibited good sphericity since it could avoid the in fl uence of cross-linker agent during the nucleation period.
MicroPCMs, suspension-like polymerization, polyvinylpyrrolidone, polystyrene-divinylbenzene, crosslinkerpostaddition
1 Introduction
In recent years it is well-known that thermal energy storage systems would provide the potential to attain energy savings, which could reduce the environmental impact related to energy use. Phase change materials (PCMs) which can absorb or release a large amount of heat during their phase transition are “latent” energy storage materials[1-2]. However, leakage of the melted PCMs during the phase change process and reactivity with the outside environment has limited their application[3]. Encapsulation of PCMs can overcome the above problems effectively. The most popular kinds of encapsulated PCMs are paraffi n[1,4-5]and n-alkanes (n-hexadecane[6-7],n-heptadecane[8-9],n-octadecane[10-11],n-eicosane[12-13]) because of their high latent heat of fusion and low price[14]. In this work, the paraf fi n with a melting point of about 55 ℃ was microencapsulated.
Interfacial polymerization[13,15], in situ polymerization[16], emulsion and suspension-like polymerization[1,5,9,12]are major chemical methods adopted thanks to their excellent properties and wide applications. As it is mentioned previously, the suspension-like polymerization has been used in this work since it is a simple, cheap and environmentally friendly method[17]. Among all the mentioned methods, the variety and amount of stabilizers as the significant parameters are crucial to the particle size distribution and morphology of microcapsules. The smaller particle size and narrower size distribution can provide higher surface area for heat transfer phenomena in such systems. Polyvinylpyrrolidone (PVP) as a stabilizer is widely used in microencapsulation[17-22]because of its easy dissolving and low toxicity. Luz Sánchez-Silva, et al.[19]prepared polystyrene microcapsules containing RT31 using different suspension stabilizers including PVA, PVP, Arabic gum, and gelatin. Using PVP as suspension stabilizers could obtain the spherical and regular microcapsules featuring lower average particle size with smooth surface. Jamekhorshid, et al.[22]developed polystyrene microcapsules using PVP as the stabilizer and investigated the in fl uenceon the percentage of stabilizer/styrene mass ratio (PVP/St wt.%). However, there is a variety of PVP with different molecular weights and most of the existing reports do not note the type of PVP clearly. To the best of our knowledge, there is still no such research that has investigated the in fl uence of different molecular weights and amounts of PVP on the morphology, particle size distribution and thermal properties of MicroPCMs.
In addition, divinylbenzene (DVB) functioning as crosslinking agents in the dispersion polymerization of styrene can improve mechanical strength and thermal stability of polymer particles. However, small amounts of DVB could broaden the particle size distribution and would lead to irregular particles. Certain articles illustrated that many of these problems could be avoided if one delayed the addition of the cross-linking agent until the end of the nucleation stage of the reaction[23-24]. The similar phenomenon takes place on the St-DVB MicroPCMs. There were many concaves on the surfaces of MicroC18 using St-DVB as the wall materials as reported by You, et al[18]. The formation of concaves might be resulted from the fact that the density of monomer was smaller than that of St-DVB polymer and the volume ofn-octadecane would shrink during the transition from a melted state to the crystalline state in the cooling process. But the MicroPCMs using polystyrene as the wall materials synthesized by Sánchez L, et al.[21]had less concaves than the MicroC18 prepared by You, et al[18]. In consequence, we conjectured that the time of adding DVB could in fl uence the morphology of St-DVB MicroPCMs as well as the St-DVB particles.
This work presented and discussed the microencapsulation of paraf fi n with a melting point of 55 ℃ in P(St-DVB) using the suspension-like polymerization method. The structure and thermal properties of the prepared MicroPCMs were characterized. The in fl uence of different molecular weights and amounts of PVP on the morphology, particle size distribution and thermal properties of MicroPCMs was investigated in detail to optimize a stabilizer. And it was for the fi rst time using the way of delaying the addition of the cross-linking agent (DVB) to improve the morphology of MicroPCMs.
2 Experimental
2.1 Materials
The materials used in experiments included the following: styrene (St, CP, provided by the Tianjin Chemical Reagent Factory) which should be washed with sodium hydroxide to remove the inhibitor, divinylbenzene (DVB, with a purity of 80%, provided by the Aladdin Reagent Chemical Co., Ltd.), paraf fi n (with a latent heat of melting of 158.6 J/g and a melting point of 55 ℃, provided by the Shanghai Huashen Rehabilitation Equipment Co., Ltd.), 2,2-azo-bisisobutyronitrile (AIBN, AR, provided by the Tianjin Kermel Chemical Reagent Co., Ltd.), and polyvinylpyrrolidone (PVP, MW 8000—130000, provided by the Aladdin Reagent Chemical Co., Ltd.).
2.2 Synthesis of MicroPCMs
The process for preparing MicroPCMs by suspension-like polymerization is shown in Figure 1. The synthesis system involved two phases: an oil phase and a water phase. The recipe used for MicroPCMs is shown in Table 1. The oil phase was obtained by mixing a predetermined mass ratio of paraf fi n, St, DVB, and AIBN. The water phase was obtained by dissolving PVP in distilled water. The oil phase was added into the water phase at 55 ℃. The mixture was emulsified at a stirring rate of 1 400 r/min for 10 min. Then the temperature was elevated to 80 ℃, and the mixture was stirred continuously at a rate of 1 000 r/min for 5 h. The resultants were washed with hot water and ethyl alcohol three times, respectively. And then MicroPCMs were dried in an oven at about 50 ℃ for 24 h prior to being collected for testing.

Table 1 The recipe used for preparation of MicroPCMs
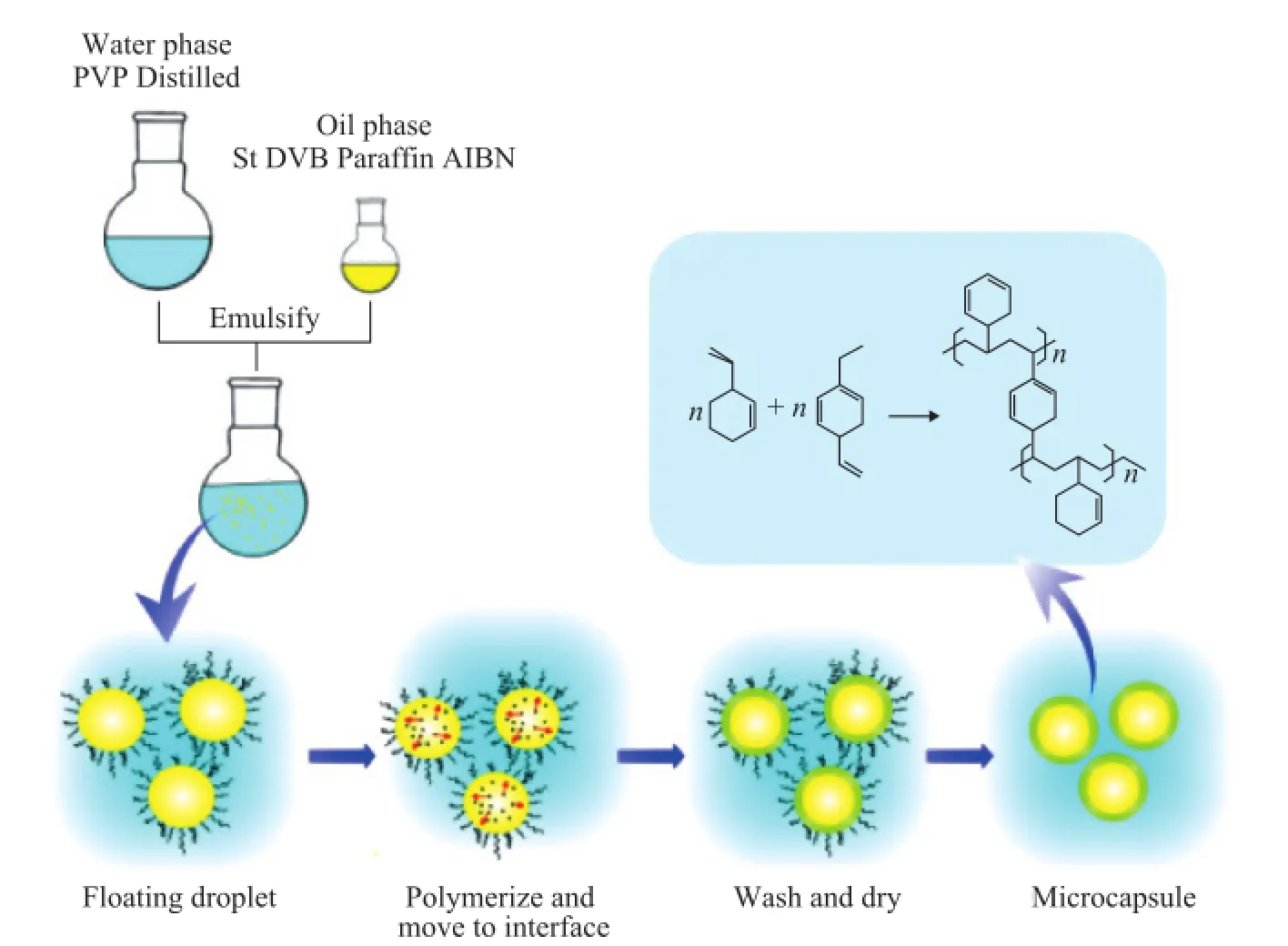
Figure 1 The process for preparing MicroPCMs
2.3 Characterization of MicroPCMs
FTIR of paraf fi n, St-DVB polymer and MicroPCMs was obtained using a Fourier transform infrared spectrometer (FTIR, TENSOR 27, Brooke Spectral Instruments) at room temperature.
The surface morphology of MicroPCMs was obtained by using a scanning electron microscope (SEM, Quanta-2000). The MicroPCMs were spread on a stainless steel SEM stub and air-dried overnight. Then the samples were coated with gold and observed with a secondary electron detector.
The particle size distribution of MicroPCMs was measured by a laser diffraction particle size analyzer (BT-9300Z). In such analyses, some of MicroPCMs were dispersed in the deionised water. The water bath was equipped with an ultrasonic probe to prevent particles from agglomeration and a pump arm to circulate the suspended microcapsules into the measuring cell.
The phase change properties of MicroPCMs, St-DVB polymer and pure paraffin were determined using a differential scanning calorimeter (DSC-60 DAOJIN). The measurements were performed by varying the temperature from 30 ℃ to 75 ℃ at a heating rate of 10 ℃/min in a nitrogen atmosphere. The ratio of encapsulated paraf fi n to St-DVB polymer can be determined using DSC results from the following equation[25].

where ΔHm,MicroPCMs(J/g) is the latent heat of the microcapsule containing paraf fi n and ΔHm,PCMs(J/g) is the latent heat of pure paraffin. In Eq. (1), it is assumed that the latent heat of the microcapsule without paraffin is zero, which is true if the shell does not undergo phase change. The thermal stability of pure paraf fi n, PS-DVB polymer and MicroPCMs was analyzed by thermogravimetry (TGDTA 6200, SEIKO INS). Analyses were performed by raising the temperature from room temperature to 600 ℃at a heating rate of 10 ℃/min in a nitrogen atmosphere.
3 Results and Discussion
3.1 Microcapsules containing paraf fi n
The FTIR spectra of paraf fi n, St-DVB polymer and MicroPCMs are shown in Figure 2. The multiple strong absorption peaks located at approximately 2 852 cm-1and 2 917 cm-1in the spectra of paraf fi n and MicroPCMs were associated with the aliphatic C—H stretching vibration. The peak at 1 472 cm-1, which was associated with the C—H bending, was also characteristic of paraf fi n. The peak at approximate 719 cm-1in the spectra of paraffinand MicroPCMs was associated with the in-plane rocking vibration of the CH2group. None of these speci fi c peaks were found in the spectra of the polymer shell. It is indicated that paraf fi n and St-DVB polymer exist as physical mixtures in MicroPCMs.

Figure 2 The FTIR spectra of paraf fi n, St-DVB polymer and MicroPCMs
The DSC curves of paraffin, St-DVB polymer and MicroPCMs are shown in Figure 3. There were two endothermic peaks of paraffin and MicroPCMs in the range of 25—70 ℃. The fi rst endothermic peak was small and wide in the scope of 25—45 ℃ caused by solid-solid phase change of paraf fi n. The other one was a principal endothermic peak attributed to solid-liquid phase change of paraffin. The melting phase change temperature of MicroPCMs was 56.1 ℃, which was a little higher than that of pure paraf fi n (55.5 ℃). This difference may be caused by the low thermal conductivity of the shell materials, which can affect the latent heat transfer rate from the outside to the inside. Therefore it can influence the phase change temperature of the encapsulated paraf fi n[26].

Figure 3 The DSC of paraf fi n, St-DVB polymer and MicroPCMs
The latent heat of melting was found to be -158.6 J/g for paraffin and -89.8 J/g for MicroPCMs. The ratio of encapsulated paraf fi n was 56.6% calculated by equation (1) because no peak of St-DVB co-polymer was watched in the same temperature range in the DSC curves.
The TG curves of paraf fi n, St-DVB polymer and MicroPCMs are shown in Figure 4. The onset decomposition temperature of paraf fi n was 200.2 ℃, and St-DVB polymer had a single decomposition temperature at 361.5 ℃. Meanwhile, MicroPCMs lost weight in two stages. The first stage of decomposition recorded a lost weight of 57.0% at temperature between 231.6 ℃ and 331.4 ℃ on account of the evaporation of paraffin. The first decomposition of MicroPCMs was by 31.4 ℃ higher than that of pure paraf fi n. It was indicated that the leakage of the core materials from the capsules was prevented and the washing step went suf fi ciently and core materials were not left on the outside of the St-DVB polymer shells[27]. The second stage of weight loss occurred between 383.1 ℃and 470.2 ℃ (43.0%) due to decomposition of St-DVB polymer. The TG results were consistent with the DSC results in terms of their weight loss.

Figure 4 The TG of paraf fi n, St-DVB polymer and MicroPCMs
In conclusion, it was confirmed that MicroPCMs were successfully compounded, and meanwhile paraffin had been successfully encapsulated by the shell of St-DVB polymer on the basis of the above-mentioned analysis.
3.2 Effect of different types of PVP on particle size distribution of MicroPCMs
The type and amount of stabilizers are the major factors in fl uencing the emulsi fi cation step in the process of Mi-croPCMs preparation. The amount and kind of stabilizers will change the interfacial tension between the oil and aqueous phase and form a thick protective interfacial fi lm by stereo-hindrance effect that can prevent droplets contact and (re-)coalescence. Hence, the amount and molecular weight of PVP are essential to the morphology, particle size distribution and core content. To investigate this dependence, experiments using different amounts of PVP with different molecular weights as the stabilizer were conducted.
Three types of PVP with different molecular weights serving as the stabilizer had been used to disperse paraf fi n in water and prepare MicroPCMs. The data given in Table 2 are the mean diameter of sample and the energy storage capacity of MicroPCMs prepared with different stabilizers. According to Table 2, MicroPCMs were successfully synthesized with 7% of PVP k-16~18 (MW 8000), while MicroPCMs using 4%—6% of PVP k-16~18 as the stabilizer failed to meet the requirement because of excessive agglomeration. The mean particle size (D50) of sample A-7 was 152.1 μm and the paraffin content was 43.5%. This fact could be related to the viscosity of the continuous phase. When MicroPCMs were prepared using PVP k-16~18, the viscosity of the aqueous phase was too low to prevent and slow down the fl occulation and (re-)coalescence between dispersed droplets. Although a concentration equating to 7 percent of PVP k-16~18 could develop MicroPCMs, the particle size of MicroPCMs was large and the core content was low. Therefore PVP k-16~18 is not the optimal stabilizer for synthesizing MicroPCMs.
Table 2 shows that four levels of PVP k-30 (MW 40000) dosage had been used in samples B-2 (2%), B-3 (3%), B-4 (4%) and B-5 (5%), with the corresponding particle size distribution being exhibited in Figure 5 (a). In the sample B-2, its particle size could not be measured because of excessive agglomeration of the MicroPCMs. There was a unimodal distribution of the samples B-3 and B-4. With 4% of PVP k-30, not only the mean particle size of MicroPCMs decreased from 131.9 μm (in B-4) to 102.3 μm (in B-3), but also the particle size distribution looked more uniform. Although the mean particle size of the sample B-5 was lower than that of the sample B-4, two peaked regions were observed in the sample B-5. The particle size distribution of MicroPCMs prepared using PVP k-88~96 (MW 130000) is shown in Figure 5 (b). The tendency of particle size using PVP k-88~96 as the stabilizer was similar to that of using PVP k-30. The mean particle size decreased gradually with the increase in the concentration of PVP k-88~96 and the particle size distribution of the sample C-3 was the narrowest.

Table 2D50and thermal properties of MicroPCMs using PVP with different molecular weights and amounts as stabilizer

Figure 5 The particle size distribution for MicroPCMs with different amount of PVP k-30 (a) and PVP k-88~96 (b)
It is speculated that PVP can provide a macromolecular barrier against the destabilizing mechanism. The more stabilizers are added during the polymerization, the smaller MicroPCMs would be generated, because a higher amount of PVP could stabilize a bigger total surface area along with generating much smaller particles. However, when the concentration of PVP is low (in samples B-3 and C-2), the particle size distribution of MicroPCMs is broad. Because the amount of PVP is not enough to form an effective and stable steric hindrance layer to make oil phase droplets be independent of each other, the segment coagulation can occur. When the amount of PVP is beyond the best quantity (in samples B-5 and C-4), theD50decreases slightly while the particle size distribution broadens obviously and even brings out multiple peaks. It is supposed that the excessive PVP cannot develop effective and stable steric hindrance layer but can also build bridges with other particles or droplets which may increase the risk of collisions between the oil phase droplets. Thereby the coagulation can take place.
The particle size distribution and SEM images of synthesized MicroPCMs with PVP k-30 and PVP k-88~96 at the same dosage (3%) are shown in Figure 6. With an increase in the molecular weight of PVP, not only the mean diameter of MicroPCMs obviously decreased from 161.5 μm (in the sample B-3) to 32.3 μm (in the sample B-4), but also the particle size distribution looked more uniform as shown in Figure 6. Figure 6, upon comparing the samples B-3 and C-3, indicates that by increasing the molecular weight of PVP from 40000 to 130000, the particle size of MicroPCMs would decrease signi fi cantly. According to the Stokes–Einstein equation, the viscosity of continuous phase and the size of droplets have an inverse relationship. In other words, stabilizers can provide a macromolecular steric hindrance effect on slowing down the fl occulation and (re-)coalescence between dispersed droplets by increasing the viscosity of the aqueous phase[28-29]. These effects can control the particle size as well as the particle size distribution of polymer particles. Correspondingly, the viscosity of the aqueous solution of PVP increases with an increasing molecular weight of PVP. As a result, PVP k-88~96 with high molecular weight used as the stabilizer can decrease the mean particle size and improve the particle size distribution of MicroPCMs as compared to the case of PVP k-30. Therefore, these results indicate that the type of PVP is one of the crucial factors in fl uencing the control of particle size in suspension polymerization.
Thermal storage energy capacities of MicroPCMs using PVP with different molecular weights are summarized in Table 2. The thermal storage energy of MicroPCMs using PVP k-16~18 as the stabilizer was relatively low than that of MicroPCMs using PVP k-30 and PVP k-88~96 as stabilizers. The thermal storage energy capacity in these experiments using PVP k-30 and PVP k-88~96 as stabilizers was similar and ranged from 49.8%—58.5%, and the highest value was equal to 58.5% corresponding to MicroPCMs which used 4% of PVP k-30 as the stabilizer. Upon considering the above results, PVP with different molecular weights as the stabilizer has little in fl uence on PCM encapsulation during the preparation of MicroPCMs.
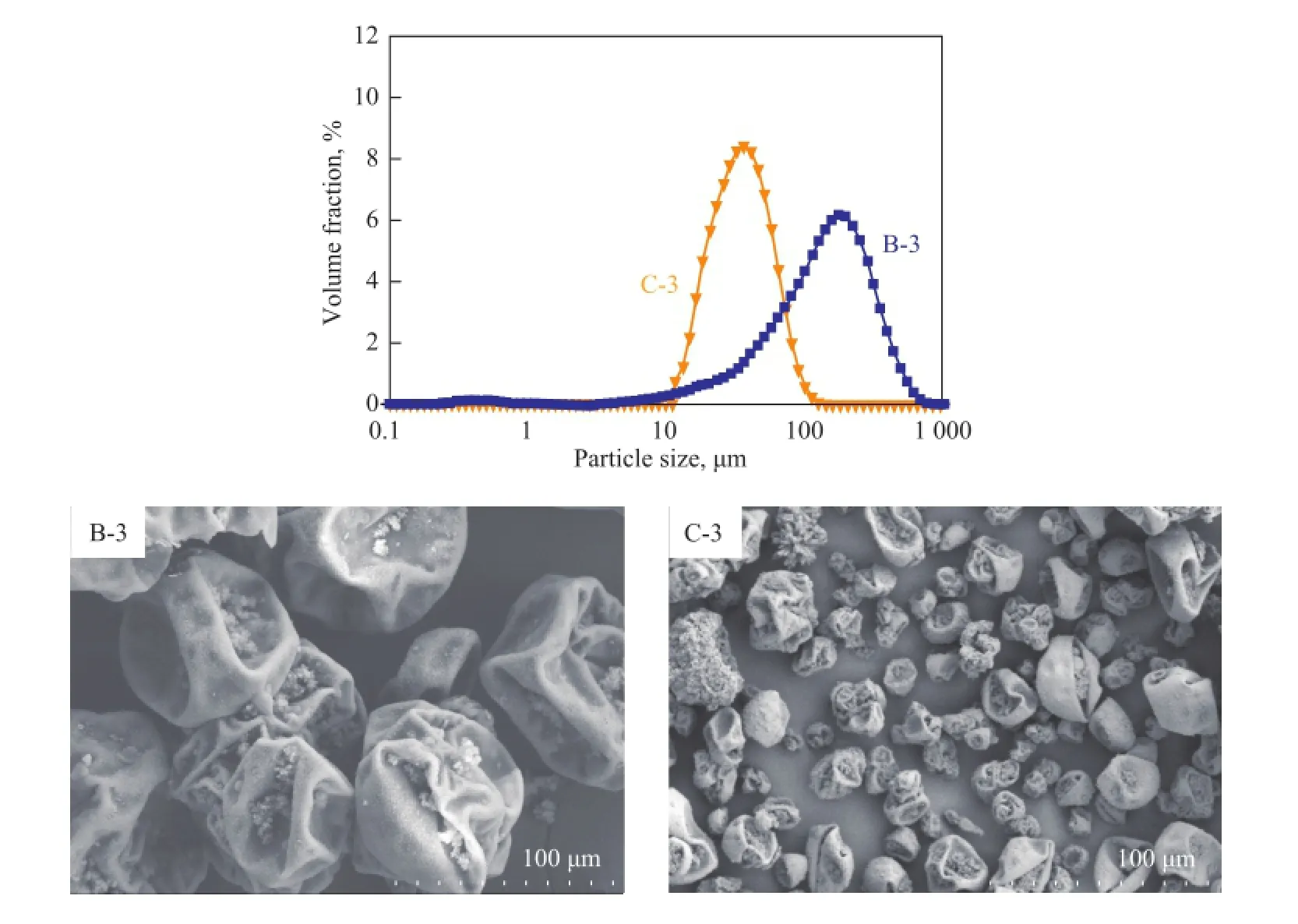
Figure 6 The particle size distribution and SEM images of MicroPCMs with different types of PVP used as stabilizer
3.3 Effect of cross-linking-postaddition on the morphology and thermal properties of microcapsules
The cross-linking-postaddition method was adopted through a slow addition of a cross-linking agent at a certain time after the start of the reaction. The recipe and procedure were the same as that described above, except that the DVB/ethanol (4:1) which was added to the sample D-3 in the 1sthour of reaction and was added to the sample E-3 in the 2ndhour of reaction after the beginning of the polymerization.
Figure 7 shows the SEM images of MicroPCMs synthesized by the crosslinker-postaddition method. DVB was added at the beginning of the reaction as shown in Figure 7 (a), and most of the MicroPCMs were seriously irregular and abnormal. The SEM image of sample D-3, to which DVB was added after 1 h of polymerization, is shown in Figure 7 (b). It can be seen that the MicroPCMs surface still had concaves, but there was a remarkable improvement as compared to Figure 7 (a). Figure 7 (c) describes the sample E-3 to which DVB was added after 2 h of polymerization. MicroPCMs exhibited good sphericity but had some little wrinklings on the surface. To sum up, the morphology of MicroPCMs was improved gradually by the crosslinker-postaddition method. It is surmised that at the beginning of the process, the St monomer, initiator and paraffin exist homogeneously in the dispersed phase. At the reaction temperature, the St monomer forms oligomer radicals. With the increase of the chain length of oligomer, the oligomers are then precipitated to form the primary particles. Meanwhile, the solubility of primary particles in oil phase decreases. Therefore, the primary particles move to oil-water interface step by step. After primary polymerization, the DVB is added to the system. The DVB either reacts with the remaining radicals in the solution or is absorbed by the primary particles. Then, the crosslinked primary particles would grow and form the capsule wall. This is proposed on the basis of this work and the published literature[24]. In the conventional encapsulation process, DVB and styrene after having been added to oil phase can take part in the polymerization reaction simultaneously. But the reaction rate of DVB is faster than that of styrene during the nucleation period of polymerization due to the difference of the numbers of reactive groups[30]. In this case, the crosslinking degree of polystyrene is greatly low while the cross-linked structure is uneven in the later stage of reaction. Therefore thecross-linking agent DVB is unevenly distributed on the surface of MicroPCMs and can limit the growth of the MicroPCMs. So MicroPCMs have seriously irregular and abnormal concaves. The crosslinker-postaddition method can avoid the influence of cross-linker agent during the nucleation period. Thus, the MicroPCMs exhibited good sphericity. But some little wrinkles can be seen on the surface of sample E-3, because the mechanical strength of St-DVB cannot resist shrinkage due to the different density between the monomer and St-DVB co-polymer and the volume ofn-octadecane would shrink during its transition from the melted state to the crystalline state during the cooling process.

Figure 7 SEM for MicroPCMs prepared by the crosslinker-postaddition method
The thermal stability of MicroPCMs is shown in Figure 8. MicroPCMs lost its weight in two stages. The fi rst decomposition temperature of MicroPCMs with different time of crosslinker addition was similar, which was higher by about 30 ℃ than the decomposition temperature of paraffin. The second decomposition temperature of MicroPCMs was around 380 ℃. This showed that MicroPCMs were successfully prepared using the crosslinkerpostaddition method.
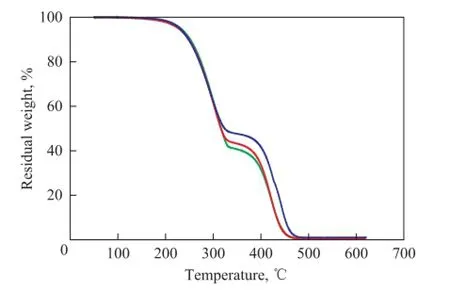
Figure 8 TG of MicroPCMs prepared by the crosslinkerpostaddition method
4 Conclusions
The MicroPCMs of paraffin with a melting point of 55℃ serving as the novel phase change material using P(St-DVB) as the shell were prepared through the suspensionlike polymerization. The characterization of samples by DSC confirmed that St-DVB/paraffin MicroPCMs were successfully synthesized. The laser particle size distribution and SEM results indicated that the variety and amount of PVP had great in fl uence on MicroPCMs. The higher molecular weight of PVP is bene fi cial to preparing smaller particle diameter of MicroPCMs. The MicroPCMs using 3% of PVP k-88~96 was optimum thanks to its high thermal storage energy (89.9 J/g) and low average diameter (32.3 μm). The morphology of MicroPCMs with DVB added in the 2ndhour of reaction is ideal except for some little wrinklings on the surface. After having grappled all the issues, a series of the P(St-DVB) / paraffin MicroPCMs with different particle sizes and morphology were prepared successfully.
Acknowledgements: This work was financially supported by the National Natural Science Foundation of China (No. 20973022 and 11472048) and the State Key Laboratory of Catalytic Materials and Reaction Engineering (RIPP, SINOPEC).
[1] Al-Shannaq R, Farid M, Al-Muhtaseb S, et al. Emulsion stability and cross-linking of PMMA microcapsules containing phase change materials[J]. Solar Energy Materials and Solar Cells, 2015, 132: 311-318
[2] Zhao C Y, Zhang G H. Review on microencapsulated phase change materials (MEPCMs): Fabrication, characterization and applications[J]. Renewable and Sustainable Energy Reviews, 2011, 15(8): 3813-3832
[3] Jamekhorshid A, Sadrameli S M, Farid M. A review of mi-croencapsulation methods of phase change materials (PCMs) as a thermal energy storage (TES) medium[J]. Renewable and Sustainable Energy Reviews, 2014, 31: 531-542
[4] Sánchez-Silva L, Rodríguez J F, Romero A, et al. Preparation of coated thermo-regulating textiles using Rubitherm-RT31 microcapsules[J]. Journal of Applied Polymer Science, 2011, 124: 4809-4818
[5] Sánchez -Silva L, Rodríguez J F, Carmona M, et al. Thermal and morphological stability of polystyrene microcapsules containing phase-change materials[J]. Journal of Applied Polymer Science, 2011, 120(1): 291-297
[6] Yin D, Liu J, Geng W, et al. Microencapsulation of hexadecane by surface-initiated atom transfer radical polymerization on a Pickering stabilizer[J]. New J Chem, 2015, 39(1): 85-89
[7] Shirin-Abadi A R, Mahdavian A R, Khoee S. New Approach for the Elucidation of PCM Nanocapsules through Miniemulsion Polymerization with an Acrylic Shell[J]. Macromolecules, 2011, 44(18): 7405-7414
[8] Sarı A, Alkan C, Kahraman Döğüşcü D, et al. Micro/nanoencapsulatedn-heptadecane with polystyrene shell for latent heat thermal energy storage[J]. Solar Energy Materials and Solar Cells, 2014, 126: 42-50
[9] Sarı A, Alkan C, Karaipekli A. Preparation, characterization and thermal properties of PMMA/n-heptadecane microcapsules as novel solid–liquid microPCM for thermal energy storage[J]. Applied Energy, 2010, 87(5): 1529-1534
[10] Wang H, Wang JP, Wang X, et al. Preparation and properties of microencapsulated phase change materials containing two-phase core materials[J]. Industrial & Engineering Chemistry Research, 2013, 52(41): 14706-14712
[11] Tang X, Li W, Zhang X, et al. Fabrication and characterization of microencapsulated phase change material with low supercooling for thermal energy storage[J]. Energy, 2014, 68: 160-166
[12] Alkan C, Sarı A, Karaipekli A. Preparation, thermal properties and thermal reliability of microencapsulatedn-eicosane as novel phase change material for thermal energy storage[J]. Energy Conversion and Management, 2011, 52(1): 687-692
[13] Shin Y, Yoo D I, Son K. Development of thermoregulating textile materials with microencapsulated phase change materials (PCM). II. Preparation and application of PCM microcapsules[J]. Journal of Applied Polymer Science, 2005, 96(6): 2005-2010
[14] Sharma A, Tyagi V V, Chen C R, et al. Review on thermal energy storage with phase change materials and applications[J]. Renewable and Sustainable Energy Reviews, 2009, 13(2): 318-345
[15] Khakzad F, Alinejad Z, Shirin-Abadi A R, et al. Optimization of parameters in preparation of PCM microcapsules based on melamine formaldehyde through dispersion polymerization[J]. Colloid and Polymer Science, 2013, 292(2): 355-368
[16] Ma Y, Chu X, Tang G, et al. The effect of different soft segments on the formation and properties of binary core microencapsulated phase change materials with polyurea/ polyurethane double shell[J]. Journal of Colloid and Interface Science, 2013, 392: 407-414
[17] Luz Sánchez E L, Carmona M, Rodríguez J F, et al. Applying an experimental design to improve the characteristics of microcapsules containing phase change materials for fabric uses[J]. Ind Eng Chem Res, 2008, 47: 9783-9790
[18] You M, Wang X, Zhang X, et al. Microencapsulatedn-octadecane with styrene-divinybenzene co-polymer shells[J]. Journal of Polymer Research, 2010, 18(1): 49-58
[19] Sánchez-Silva L, Rodríguez J F, Sánchez P. Influence of different suspension stabilizers on the preparation of Rubitherm RT31 microcapsules[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2011, 390(1-3): 62-66
[20] Sánchez-Silva L, Rodríguez J F, Romero A, et al. Microencapsulation of PCMs with a styrene-methyl methacrylate copolymer shell by suspension-like polymerisation[J]. Chemical Engineering Journal, 2010, 157(1): 216-222
[21] Sánchez L, Sánchez P, Carmona M, et al. In fl uence of operation conditions on the microencapsulation of PCMs by means of suspension-like polymerization[J]. Colloid and Polymer Science, 2008, 286(8/9): 1019-1027
[22] Jamekhorshid A, Sadrameli S M, Bahramian A R. Process optimization and modeling of microencapsulated phase change material using response surface methodology[J]. Applied Thermal Engineering, 2014, 70(1): 183-189
[23] Song J S, Winnik M A. Cross-linked, monodisperse, micron-sized polystyrene particles by two-stage dispersion polymerization[J]. Macromolecules 2005, 38(20): 8300-8307
[24] Cui H, Chen H, Qu R, et al. Synthesis of monodispersecrosslinked polystyrene microspheres via dispersion copolymerization with the crosslinker-postaddition method[J]. Journal of Applied Polymer Science, 2008, 107(6): 3909-3916
[25] Sa′nchez-Silva L, Tsavalas J, Sundberg D, et al. Synthesis and characterization of paraffin wax microcapsules with acrylic-based polymer shells[J]. Ind Eng Chem Res, 2010, 49(23): 12204-12211
[26] Salaun F, Devaux E, Bourbigot S, et al. Development of phase change materials in clothing. Part I: Formulation of microencapsulated phase change materials[J]. Textile Research Journal, 2009, 80(3): 195-205
[27] Ma Y, Sun S, Li J, et al. Preparation and thermal reliability of microencapsulated phase change materials with binary cores and acrylate-based polymer shells[J]. Thermochimica Acta, 2014, 588: 38-46
[28] Hwang J S, Kim J N, Wee Y J, et al. Factors affecting the characteristics of melamine resin microcapsules containing fragrant oils[J]. Biotechnology and Bioprocess Engineering, 2006(5): 391-395
[29] Seid M J, Beheshtib P, Assadpoorc E. Rheological behavior and stability of d-limonene emulsions made by a novel hydrocolloid (Angum gum) compared with Arabic gum[J]. Journal of Food Engineering, 2012(1): 1-8
[30] Lee K C, Wi H A. Highly crosslinked micron-sized monodispersed polystyrene particles by semicontinuous dispersion[J]. Journal of Applied Polymer Science, 2009, 115(5): 3092-3102
Received date: 2015-10-23; Accepted date: 2016-01-07.
Prof. Zhao Tianbo, Telephone: +86-13522293506; E-mail: zhaotb@bit.edu.cn.
杂志排行

中国炼油与石油化工的其它文章
- Experimental Study of UDS Solvents for Purifying Highly Sour Natural Gas at Industrial Side-stream Plant
- Highly Active and Stable Ni2P/SiO2Catalyst for Hydrogenation of C9Petroleum Resin
- Enhanced Performance of Denitrifying Sul fi de Removal Process by 1,2-Naphthoquinone-4-Sulphonate
- Investigation of Different Coke Samples Adhering to Cyclone Walls of a Commercial RFCC Reactor
- Numerical Study of Air Nozzles on Mild Combustion for Application to Forward Flow Furnace
- Preparation of Core-Shell Composite of Y@Mesoporous Alumina and Its Application in Heavy Oil Cracking