N末端Strep蛋白标签表达载体的构建和应用
2016-03-21吉宏张包小峰
石 禹,吉宏张,包小峰
(南通大学1.附属第三人民医院临床药学室、2.药学院,江苏南通 226006)
N末端Strep蛋白标签表达载体的构建和应用
石禹1,2,吉宏张2,包小峰2
(南通大学1.附属第三人民医院临床药学室、2.药学院,江苏南通226006)
中国图书分类号: R341; R345.57; R374; R394.2; R977. 6
摘要:目的构建携带N末端Strep( NS)蛋白标签的表达载体;在表达载体中构建衣原体RNA聚合酶亚基重组蛋白并表达。方法采用PCR的方法,通过引物引入NS蛋白标签和新的多克隆位点替代pET21c-DH质粒中原有的T7蛋白标签和多克隆位点,选择新引入的Not I酶切位点进行PCR产物的环化自连,转化DH-5α细菌后筛选阳性菌株,PCR法和基因测序法鉴定新构建的表达载体;在新构建表达载体的BamH I和Sal I位点之间分别插入衣原体RNA聚合酶核心酶的α、β、β'亚基,获得表达NS-α、NS-β、NS-β'融合蛋白的表达载体,转化表达菌株ArcticExpress,筛选阳性表达菌株,并用考马斯亮蓝染色、Western blot等法鉴定融合蛋白的表达情况。结果成功构建了携带NS蛋白标签的pET21c-NSMCS载体,并成功将其应用于衣原体RNA聚合酶核心酶亚基融合蛋白的构建及表达。结论获得稳定表达NS-α、NS-β、NS-β'融合蛋白的表达载体,为研究衣原体基因转录调控奠定了良好的基础。
关键词:Strep蛋白标签;表达载体;融合蛋白;克隆; RNA聚合酶;衣原体
包小峰( 1979-),男,博士,教授,硕士生导师,研究方向:衣原体转录调控与抗衣原体化合物,通讯作者,E-mail: baoxi@ ntu.edu.cn
蛋白标签是指利用DNA体外重组技术与目标蛋白基因融合表达的一种多肽或蛋白,常用于目标蛋白的构建、表达、检测、示踪及纯化[1]。在重组蛋白研究过程中,通常使用亲和性标签(如His、GST、Strep)等。主要因这些亲和标签能使重组蛋白的纯化、检测、固定等变得更方便易行。同时,蛋白的表达和纯化都可以实现规模化[2]。本文主要介绍蛋白质N末端Strep标签表达载体的构建以及其在研究衣原体RNA聚合酶核心酶中的应用。
1 材料
1.1质粒、菌株质粒pET21c-DH( His标签被去除的pET21c,美国罗格斯-新泽西州立大学的范辉宙教授惠赠),DH-5α化学感受态细胞购自于南京诺唯赞生物科技有限公司,ArcticExpress表达菌株的化学感受态细胞购自美国Stratagene公司。
1.2工具酶与主要试剂限制性内切酶、T4连接酶购自NEB(北京)有限公司;高保真Phanta DNA聚合酶、ClonExpress II非连接依赖型快速克隆试剂盒购自南京诺唯赞生物科技有限公司; SanPrep柱式质粒DNA小量抽提试剂盒、SanPrep柱式DNA胶回收试剂盒、SanPrep柱式PCR产物纯化试剂盒、异丙基硫代半乳糖苷( IPTG)均购自上海生工生物工程有限公司; 2×Taq PCR MasterMix为天根生化科技(北京)有限公司产品。
2 N末端Strep蛋白标签表达载体的构建
2.1引物设计与合成根据pET21c-DH质粒载体的序列,采用Primer Premier 6. 0软件进行PCR引物设计,插入Strep标签的同时将pET21c-DH( Fig 1A)中原有的多克隆位点( multiple cloning sites,MCS)替换为( Fig 1B)所示的多克隆位点,预计得到产物pET21c-NS-MCS质粒。引物由上海生工生物工程股份有限公司合成,引物序列如下,上游引物: 5'-TATATGCGGCCGCAATTGAATTCCTCGAGTGAGATC CGGCTGCTAAC-3';下游引物:5'-TATATGCGGCCGCGTCGACCATGGATCCTTTTTCGAACTG-3'。其中GCGGCCGC为环化连接PCR产物所需的Not I酶切位点。按照以下体系进行PCR反应: 200 nmol ·L-1上游引物、200 nmol·L-1下游引物、30 ng pET21c-DH模板质粒、0. 2 U Phanta DNA聚合酶,ddH2O补足至50 μL;在PCR仪上按照如下反应条件进行扩增: 95℃预变性3 min,95℃变性15 s,56℃退火15 s,72℃延伸6 min,共24个循环。得到的PCR扩增产物在琼脂糖凝胶电泳检测,条带指示约在5 400 bp( Fig 2A),为预期的MCS序列片段带。然后使用SanPrep柱式DNA胶回收试剂盒胶回收得到带有MCS序列的载体片段。
2.2酶切和连接反应将以上胶回收后的序列片段进行单酶切反应,体系如下:序列片段30 μL,10× CutSmart缓冲液5 μL,Not I限制性内切酶1 μL,加ddH2O至50 μL,37℃水浴酶切3 h。使用SanPrep柱式PCR产物纯化试剂盒对酶切产物进行回收,得到酶切序列片段。对该酶切序列片段使用T4连接酶进行自连反应,体系如下:酶切序列片段8 μL,10 ×T4连接酶缓冲液1 μL,T4连接酶1 μL,16℃连接过夜。
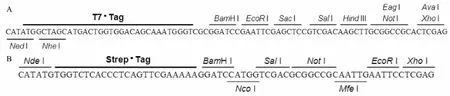
Fig 1 pET21c-DH plasmid sequence to be replaced( A) and newly introduced Strep-tag and MCS sequence in pET21c-NS-MCS Plasmid( B)
2.3重组质粒的转化、筛选和鉴定将连接产物与DH-5α化学感受态细菌混合,42℃热休克60 s后,涂布到含氨苄青霉素(终浓度为100 mg·L-1)的LB固体培养基平板上,37℃恒温箱培养14 h,挑选得到的菌落进行PCR验证,验证上游引物: 5'-GCGAAATTAATACGACTCAC-3'[pET-21c-Reverse ( 313-332)];下游引物: 5'-GGGGTTATGCTAGTTATTGC-3'[pET-21c-Forward ( 61-80)],反应酶为2× Taq PCR Master Mix共25 μL体系。在PCR仪上按照如下程序进行处理: 94℃3 min,95℃30 s,55℃30 s,72℃1 min,26个循环。PCR产物于琼脂糖凝胶电泳检测,得到约220 bp大小的条带( Fig 2B),与预期大小相符。
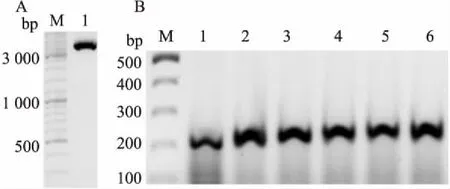
Fig 2 Vector fragment containing new MCS sequence( A) and identification of pET21c-NS-MCS clone( B) detected by agarose gel electrophoresis
2.4重组质粒的测序分析挑取经PCR鉴定的阳性菌落,接种到10 mL含100 mg·L-1氨苄青霉素的LB液体培养基中,37℃、220 r·min-1振荡培养12 h放大后送上海生工公司进行测序鉴定。测序结果显示,本实验得到的重组质粒pET21c-NS-MCS 中NS标签和多克隆位点序列和位置与预先设计的完全一致( Fig 3)。
3 N末端Strep标签表达载体在研究衣原体RNA聚合酶核心酶中的应用
衣原体是一类真核细胞内寄生,具有独特发育周期,并能通过细菌滤器的原核细胞型微生物[4]。在自然界中传播很广泛,常寄生于人类、鸟类及哺乳动物,并可引起多种疾病。其特殊的生长条件和基因转录调控所需要的遗传基础系统的缺少妨碍了对衣原体的深入研究[5]。
现代分子生物学证实转录是基因表达的第一步,也是最关键的一步,其中催化转录的RNA聚合酶在基因转录调控过程中占有重要的地位[6]。通常的RNA聚合酶是一种由多个蛋白亚基组成的复合酶,包括α、β、β'、σ等亚基。其中α亚基决定哪些基因被转录;β亚基含有三磷酸核苷的结合位点; β'亚基含有与DNA模板相结合的位点;σ亚基与转录起始点的识别有关[7]。通过质粒载体的研究我们已经证实GrgA是衣原体独有的转录激活因子,在衣原体生长发育周期中扮演着十分重要的角色[3]。但目前关于GrgA与衣原体RNA聚合酶关系的文献报道很少,已发现GrgA可以和衣原体DNA和RNA聚合酶的σ66亚基结合[3,8],但与RNA聚合酶核心酶亚基的关系都是未知的。本文通过构建N末端Strep标签表达载体,为我们研究GrgA与衣原体RNA聚合酶核心酶各亚基间的相互关系提供了实验基础。
3.1构建携带NS标签的衣原体RNA聚合酶的α、β和β'亚基为了提高效率,我们采用ClonExpress技术,它是一种简单、快速并且高效的DNA定向克隆技术,可将插入片段PCR产物定向克隆至任意载体的任意位点。首先根据GenBank中已收录的沙眼衣原体L2血清型来源的衣原体RNA聚合酶的α亚基基因( GenBank Accession: NC_010287. 1 903145-904278)、β亚基基因( GenBank Accession: NC_010287. 1 673171-676929)、β'亚基基因( Gen-Bank Accession: NC_010287. 1 668956-673146),由Primer Premier 6. 0软件进行PCR引物设计。α亚基引物序列如下:上游引物: BamH I-RpoA-5'f: 5'-CCTCAGTTCGAAAAAGGATCCATGTCGGATAGTTC ACACA-3';下游引物: Sal I-RpoA-3' f: 5'-TTCAATT GCGGCCGCGTCGACTTATCCCTTGGTATTTTTACTC-3'。β亚基引物序列如下:上游引物: BamH I-RpoB-5' f:5'-CCTCAGTTCGAAAAAGGATCCATGTTCAAG TGCCCGG-3';下游引物: Sal I-RpoB-3'f: 5'-TTCAATTGCGGCCGCGTCGACTTAAGCATCTACTACCATAGG G-3'。β'亚基引物序列如下:上游引物: BamHIRpoC-5' f:5'-CCTCAGTTCGAAAAAGGATCCATGTTCAGAGAAGGTTCTCG-3';下游引物: Sal I-RpoC-3' f:5'-TTCAATTGCGGCCGCGTCGAC GGCAGATCTTTAACCTG-3'。以上引物中GGATCC为BamH I识别位点; GTCGAC为Sal I识别位点;模板为沙眼衣原体L2血清型基因组DNA( genomic DNA,gDNA)。PCR反应体系与条件同上。得到的PCR扩增产物在琼脂糖凝胶电泳检测( Fig 4A),胶回收后作为插入片段。
将上述pET21c-NS-MCS质粒进行BamH I、Sal I限制性内切酶双酶切,37℃水浴酶切3 h后,将酶切产物于琼脂糖凝胶电泳分离纯化( Fig 4B),胶回收得到载体片段。
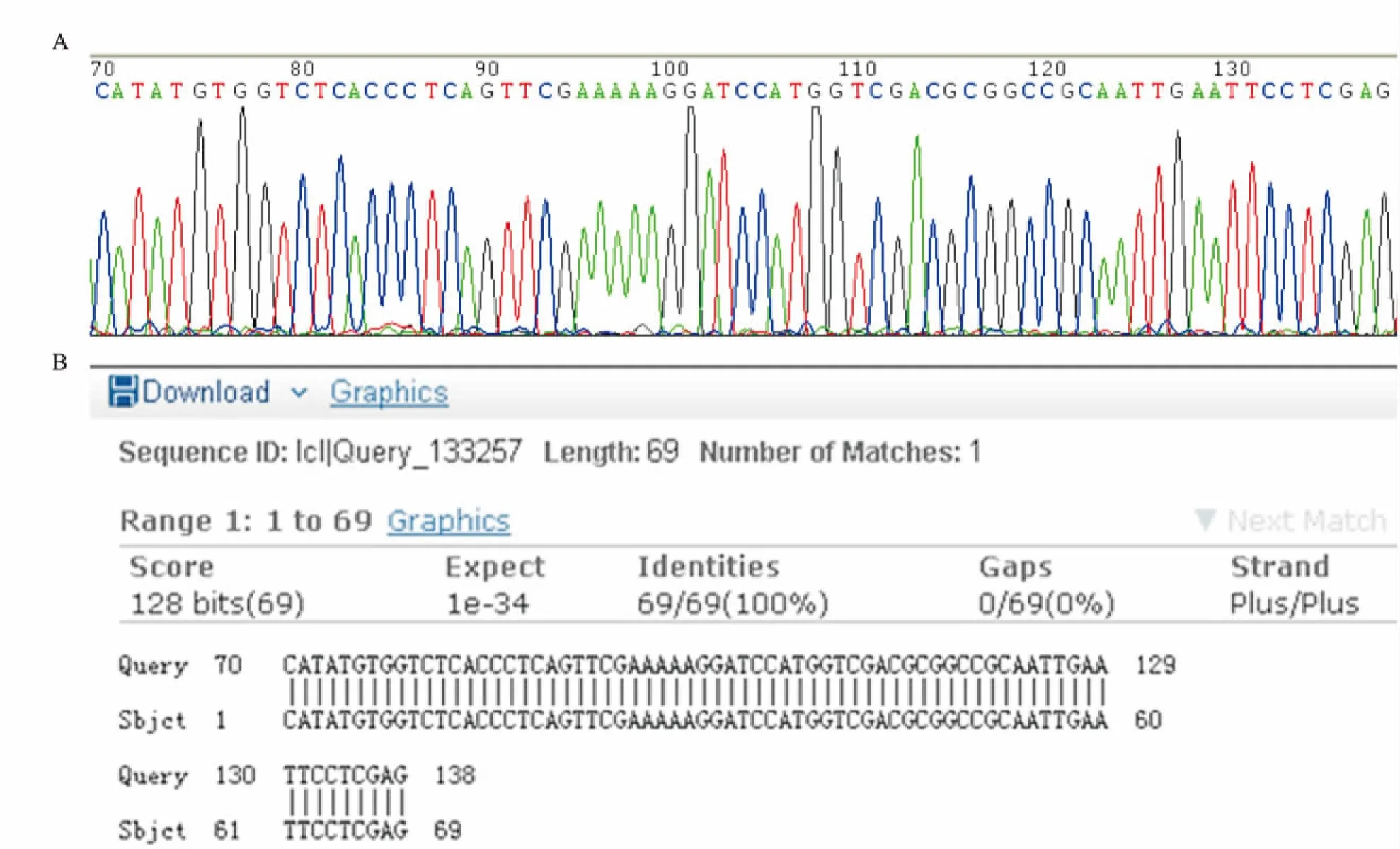
Fig 3 pET21c-NS-MCS Plasmid sequencing results( A) and sequence comparison( B)
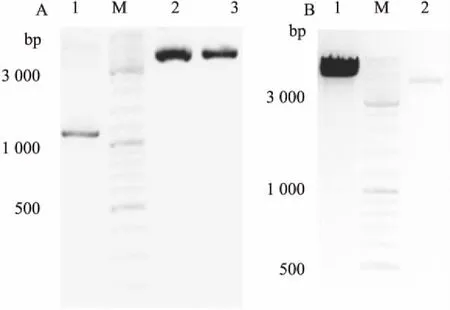
Fig 4 Insertion fragment( A) and vector fragment( B) of construction NS-α of NS-β、NS-β' detected by Agarose gel electrophoresis
将以上3个插入片段分别与载体片段按摩尔比4: 1进行混合,加入Clon-Express重组酶ExnaseTMII 37℃水浴反应30 min。将重组产物转化至DH-5α化学感受态细菌后,涂布到相应抗性的LB固体培养基平板上,37℃恒温箱培养14 h,挑选得到的菌落进行PCR验证。各挑选一株经鉴定为阳性的菌落接种放大后送测序,测序结果无突变(验证结果此处省略)。
3.2衣原体NS-α、NS-β和NS-β'融合蛋白的表达
将重组质粒分别转化到表达菌株ArcticE-xpress,涂布于含相应抗性和庆大霉素的LB固体培养基平板上,37℃培养过夜。d 2挑选阳性克隆,接种到5 mL含相应抗性的LB液体培养基中,37℃、220 r·min-1振荡培养12 h至对数成长期后按1∶100的比例取2. 5 mL至250 mL LB液体培养基中,30℃、220 r·min-1振荡培养约3 h放大至光密度( OD)为0. 8后,加入1 mmol·L-1IPTG,180 r· min-113℃低温振荡表达12 h。高速离心8 000 r· min-1收集细菌沉淀,加入含25 mmol·L-14-羟乙基哌嗪乙磺酸( Hepes)、300 mmol·L-1氯化钠的非变性细菌裂解液10 mL,超声裂解细菌后12 000 r· min-1离心收集上清,即得到目标蛋白粗样品,Western blot( Fig 5A)及考马斯亮蓝染色( Fig 5B)检测结果显示相应亚基的融合蛋白,表明携带NS标签的衣原体RNA聚合酶的α、β和β'亚基表达载体成功构建并表达。
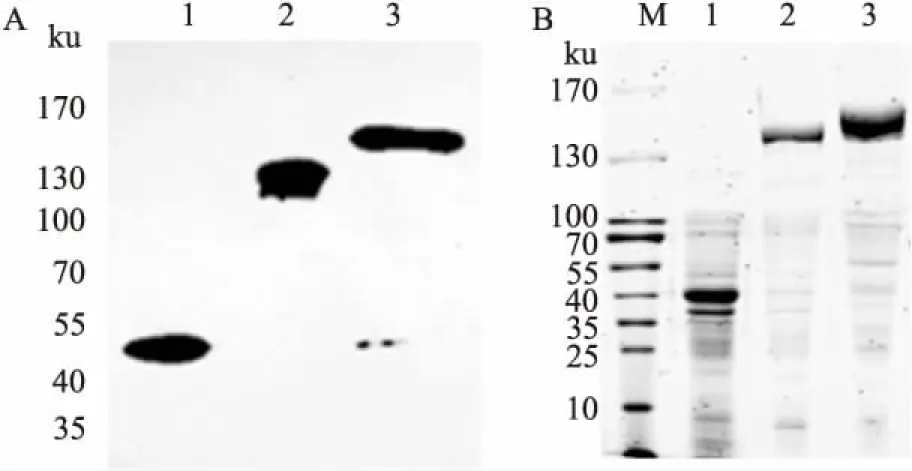
Fig 5 Western blot( A) and Coomassie bright blue staining( B) used to detect expression of NS-α,NS-β,NS-β' Fusion Protein
4 讨论
本实验应用的蛋白标签技术在检测和纯化目的蛋白、蛋白定位和转运、蛋白质相互作用以及构象改变等研究中发挥重要作用[9]。其中Strep蛋白标签由仅8个氨基酸组成,对蛋白质的结构和活性通常没有影响,同时具有高选择性和易于控制的结合性,适合从真核或原核表达体系中纯化strep标签蛋白[10]。在此基础上开发的Strep-tag/Strep-Tactin系统现已成为广泛应用的蛋白亲和层析系统之一。其可与多种不同的基质偶联,使Strep-tag融合蛋白得以在生理条件下亲和纯化。与其他tag相比,这些温和的纯化参数能保存蛋白质的生物活性,并经进一步层析后即可产出超过99%的纯度[11]。
设计pET21c-NS-MCS质粒载体中新的多克隆位点时充分考虑覆盖本研究课题所在实验室常用并已购买的限制性内切酶酶切位点,为进一步构建缺失部分序列的衣原体RNA聚合酶亚基融合蛋白提供了一个相对“多能”的中介质粒载体,同时我们在Strep蛋白标签的前面加入一个新的Nde I酶切位点( Fig 1B),这样在构建不带Strep标签甚至替换其它亲和性标签衣原体RNA聚合酶各亚基时也同样可以利用这一质粒载体[12]。另外本实验在构建pET21c-NS-MCS质粒载体时利用的是Not I限制性内切酶环化连接,新的多克隆位点Not I限制性内切酶所处位置较有利于引物的设计,而原有多克隆位点会造成引物过长,这样PCR产物的效率、目的融合蛋白的序列突变率可能会产生问题。
融合蛋白技术主要是为了获得大量标准融合蛋白而进行的有目的性的基因融合和蛋白表达方法[13]。在实验时,应该多考虑几种构建融合蛋白常用的融合方式,从中筛选出效率比较高的连接方式。比如以上我们在构建携带NS标签的衣原体RNA聚合酶的α、β和β'亚基时所采用的ClonExpress技术就明显比T4连接酶连接的方式效率高很多,具体表现在琼脂糖胶回收的产物浓度、转化感受态细菌的效率、目的克隆的阳性率等方面。本次实验很好的利用了融合蛋白和克隆技术,成功构建了N末端带有Strep标签的pET21c-NS-MCS质粒,其含有适合进一步构建融合蛋白的多克隆位点,并将其成功应用于衣原体RNA聚合酶核心酶亚基重组表达载体的构建,为我们研究GrgA与衣原体RNA聚合酶各亚基间的相互作用提供了良好的基础。
(致谢:本研究课题主要在南通大学药学院生物医药重点实验室与南通大学附属第三人民医院肝病实验室完成,感谢实验室老师和同学的帮助与支持,尤其感谢包小峰老师的细心指导与大力支持,吉宏张同学的全力协助与参与! )
参考文献:
[1]Wood D W,Camarero J A.Intein applications: from protein purification and labeling to metabolic control methods[J].J Biol Chem,2014,289( 21) : 14512-9.
[2]Mizukami S,Hori Y,Kikuchi K,et al.Small-molecule-based protein-labeling technology in live cell studies: probe-design concepts and applications[J].Acc Chem Res,2014,47( 1) : 247-56.
[3]Bao X F,Nickels B E,Fan H,et al.Chlamydia trachomatis protein GrgA activates transcription by contacting the nonconserved region of σ66[J].Proc Natl Acad Sci USA,2012,109( 42) : 16870-5.
[4]Nunes A,Gomes J P.Evolution,phylogeny,and molecular epidemiology of Chlamydia[J].Infect Gent Evol,2014,4( 23) : 49-64.
[5]Bachmann N L,Polkinghorne A,Timms P,et al.Chlamydia genomics: providing novel insights into chlamydial biology[J].Trends Microbiol,2014,22( 8) : 464-72.
[6]Mekler V,Minakhin L,Borukhov S,et al.Coupling of downstream RNA polymerase-promoter interactions with formation of catalytically competent transcription initiation complex[J].J Mol Biol,2014,426( 24) : 3973-84.
[7]Rao X,Deighan P,Hua Z,et al.A regulator from Chlamydia trachomatis modulates the activity of RNA polymerase through direct interaction with the beta subunit and the primary sigma subunit [J].Genes Dev,2009,23( 15) : 1818-29.
[8]Bao X F,Pachikara N D,Oey C B,et al.Noncoding Nucleotides and Amino Acids near the Active Site Regulate Peptide Deformylase Expression and Inhibitor Susceptibility in Chlamydia trachomatis[J].Microbiology,2011,157( Pt 9) : 2569-81.
[9]Cole N B.Site-specific protein labeling with SNAP-tags[J].Curr Protoc Protein Sci,2013,73( 30) : 1001-4.
[10]Dammeyer T,Timmis K N,Tinnefeld P.Broad host range vectors for expression of proteins with ( Twin-) Strep-tag,His-tag and engineered,export optimized yellow fluorescent protein[J].Microb Cell Fact,2013,12( 49) : 1186-97.
[11]Maertens B,Spriestersbach A,Kubicek J,et al.Strep-tagged protein purification[J].Methods Enzymol,2015,14( 72) : 53-6.
[12]陈钟琳,姜红岩,洪晓冰,等.hi FGF2真核表达载体的构建及其高表达对细胞凋亡的影响[J].中国药理学通报,2014,30 ( 11) : 1535-8.
[12]Chen Z L,Jiang H Y,Hong X B,et al.Construction of Hi FGF2 eukaryotic expression plasmids and its over-expression induced cell apoptosis[J].Chin Pharmacol Bull,2014,30( 11) : 1535-8.
[13]Janczak M,Bukowski M,Górecki A,et al.A systematic investigation of the stability of green fluorescent protein fusion proteins[J].Acta Biochim Pol,2015,62( 3) : 407-11.
Construction and application of N-terminal Strep-tagged protein expression vector
SHI Yu1,2,JI Hong-zhang2,BAO Xiao-feng2
( 1.Dept of Clinical Pharmacy,the Third People's Hospital of Nantong University,Nantong Jiangsu 226006,China; 2.School of Pharmacology,Nantong University,Nantong Jiangsu 226006,China)
Abstract:AimsTo construct the N-terminal Streptagged ( NS-tagged) fusion protein expression vector,and to apply the vector to express NS-tagged fusion proteins of Chlamydia RNA polymerase subunit.Methods By using PCR method,NS fusion protein tag and a new multiple cloning sites ( MCS) were inserted into pET21c-DH plasmid by primers to replace the original T7 protein tag and MCS.The newly introduced Not I cutting site was chosen for self-ligation of PCR product.Then,the cyclized PCR product was transformed into DH-5α competent cells.The positive clones were selected by PCR and sequencing.To get NS-tagged fusion proteins of chlamydial RNA polymerase subunits,the α,β and β' subunits were inserted between BamH I and Sal I cutting sites of the newly constructed expression vector.Then,the NS-α,NS-β and NS-β' expression vectors were transformed into ArcticExpress expression cells.The fusion protein expression statuses of transformed cells were identified by Commassie blue staining and Western blot.ResultsThe NS-tagged fusion protein expression vector pET21c-NS-MCS was successfully constructed,and NS-α,NS-β and NS-β' fusion proteins were obtained by using this newly constructed expression vector.Conclusions In this project,we constructed an NS-tagged fusion protein expression vector and applied it to express NS-α,NS-β and NS-β' fusion proteins.Our study can lay a solid foundation for the study of transcriptional regulation of Chlamydia genes.
Key words:Strep-tag; expression vector; fusion protein; cloning; RNA polymerase; chlamydia
作者简介:石禹( 1984-),男,硕士,主管药师,研究方向:抗感染临床药学,E-mail: 935748481@ qq.com;
基金项目:国家自然科学基金资助项目( No 31370209,31400165) ;南通大学研究生科研创新计划项目( No 13025166) ;“青蓝工程”资助项目
收稿日期:2015-09-29,修回日期: 2015-11-29
文献标志码:A
文章编号:1001-1978( 2016) 01-0098-05
doi:10.3969/j.issn.1001-1978.2016.01.021
