当归破壁饮片-破壁粉体-传统饮片的HPLC指纹图谱研究*
2016-03-06刘星云彭丽华成金乐
刘星云,彭丽华,成金乐*
(1. 广州中医药大学中药资源科学与工程研究中心 岭南中药资源教育部重点实验室 广州 510006;2. 国家中医药管理局中药破壁饮片技术与应用重点研究室 中山 528437;3. 中山市中智药业集团有限公司 中山 528437)
当归破壁饮片-破壁粉体-传统饮片的HPLC指纹图谱研究*
刘星云1,2,3,彭丽华2,3,成金乐2,3*
(1. 广州中医药大学中药资源科学与工程研究中心 岭南中药资源教育部重点实验室 广州 510006;2. 国家中医药管理局中药破壁饮片技术与应用重点研究室 中山 528437;3. 中山市中智药业集团有限公司 中山 528437)
目的:建立当归破壁饮片(成品)、破壁粉体(中间体)及传统饮片(原料)的HPLC指纹图谱,并测定三者的阿魏酸含量,为当归破壁饮片的质量评价提供依据。方法:采用高效液相色谱法,Aglient-ZORBAX SB色谱柱,以乙腈-1%醋酸溶液为流动相进行梯度洗脱,流速为1.0 mL·min-1,检测波长为254 nm,柱温为30℃。采用2015版《中国药典》HPLC含量测定方法测定阿魏酸含量。结果:建立了当归破壁饮片-破壁粉体-传统饮片的HPLC指纹图谱,10批当归破壁饮片、10批破壁粉体及10批传统饮片的指纹图谱共有模式中均有14个共有特征峰,各批次破壁饮片、破壁粉体及传统饮片的图谱基本一致,相似度均大于0.96;当归破壁饮片中阿魏酸含量略高于当归药材。结论:本文所建立的方法稳定、可靠、重复性好,可用于当归破壁饮片的质量控制和综合评价。
当归 破壁饮片 指纹图谱 相关性 HPLC
药材当归为伞形科植物当归Angelica sinensis(Oliv.)Diels 的干燥根,具补血、活血、调经、润肠之功效,为血家之圣药。当归药材主要含有有机酸类、挥发油类、生物碱类、有机酸类和酚类[1]等成分。当归破壁饮片是通过破细胞壁粉碎技术将当归传统饮片粉碎成破壁粉体,再制粒而成的。与传统饮片相比,破壁饮片具有成分利用率高、质量均一、应用便捷和灵活等特点[2]。由于外观性状及显微特征均已被破坏,传统饮片的质量评价标准已不适用于当归破壁饮片的质量评价,而基于整体化学表征基础上的中药HPLC指纹图谱能反映中药组分概貌,通过相似程度的比较达到鉴别真伪和判断药物批次间质量是否稳定的目的[3]。目前已有较多关于当归药材的含量测定和指纹图谱分析的相关报道[4-12],但未见其破壁饮片的相关报道。本研究同时建立破壁饮片-破壁粉体(破壁饮片制备过程的中间体)-传统饮片(破壁饮片制备的原料)的HPLC指纹图谱,分析三者的相关性,对破壁饮片成品质量及制备工艺对成分的影响进行全面评价。
1 仪器与试药
1.1 仪器
安捷伦1200 RRLC快速液相色谱仪(配有G1315C型号DAD检测器、G4218A的ELSD检测器、G1312B二元泵、G1329B自动进样器、G1322A脱气机、G1316B柱温箱,Agilent ChemStation色谱工作站),TM-D24UV明澈纯水系统,KQ-400KOE型超声波清洗器(昆山市超声仪器有限公司);DK-S24电热恒温水浴锅(上海森信实验仪器有限公司);AB204-S万分之一电子天平(METTLER TOLEOD)、AUW220D十万分之一电子天平(日本岛津公司)。
1.2 试药和试剂
当归药材共10批,经中山市中智药业集团有限公司中药师贾世清鉴定为伞形科植物当归Angelica sinensis(Oliv.)Diels 的干燥根,批号及产地信息见下表1。各批药材分别按照法定炮制工艺制成相对应批次的当归传统饮片、当归破壁粉体和当归破壁饮片(均由中山市中智药业集团有限公司制备),阿魏酸对照品(批号:110773-201313,购自中国药品生物制品检定所)。

表1 当归样品信息
2 方法与结果
2.1 色谱条件
Agilent ZORBAX SB-C18色谱柱(4.6 mm×250 mm,5 μm);流动相1%醋酸溶液(A)-乙腈(B)洗脱梯度见表2;柱温为30℃;检测波长为254 nm;进样量10 μL;流速:1.0 mL·min-1。
2.2 供试品溶液的制备
将当归药材(或当归破壁粉体、当归破壁饮片)粉碎后过60目筛,取粉末1 g,精密称定,置具塞锥形瓶中,精密加入80%甲醇20 mL,称定重量,超声提取20 min(功率500 W,频率40 kHz),放置至室温,称重,补足失重,摇匀,滤过,0.22 μm微孔滤膜滤过,备用。
2.3 对照品溶液的制备
精密称取阿魏酸适量,加甲醇配制成0.1058 mg·mL-1的对照品溶液。
2.4 方法学考察
2.4.1 专属性试验
精密吸取空白溶剂(甲醇)10 μL注入液相色谱仪,按照2.1项下色谱条件进行测定,在相应保留时间处无色谱峰,表明甲醇(空白)对测定结果无干扰。
2.4.2 精密度试验
取同一份当归破壁饮片供试品溶液(批号:UGP20130101),连续进样6次进行测定,记录图谱。各主要色谱峰的相对保留时间和相对 峰面积的RSD<1.5%(参照物为阿魏酸),表明仪器精密度良好,符合指纹图谱技术要求。
2.4.3 重复性试验
取同一批次当归破壁饮片(批号:UGP20130101))6份,按2.2项下方法平行制备供试品溶液6份,按2.1项下色谱条件进行测定,记录图谱。各主要色谱峰的相对保留时间和相对峰面积基本一致,RSD<3%(参照物阿魏酸),表明本方法的重复性符合指纹图谱控制技术的要求。
2.4.4 稳定性试验
取同一批次当归破壁饮片的供试品溶液(批号:UGP20130101),精密称定,按2.2项下方法制备供试品溶液,室温下放置。分别于0、4、8、12、24 h内测定,记录图谱。计算各图谱中主要色谱峰峰面积和相对保留时间的RSD。结果显示,各主要色谱峰峰面积和相对保留时间RSD<3.0%,表明供试品溶液在24 h内稳定性良好。
2.5 指纹图谱的建立
2.5.1 指纹图谱的采集
取10批次当归传统饮片、10批次当归破壁粉体和10批次当归破壁饮片,分别按2.2项下方法制备成供试品溶液,按2.1项下色谱条件进行分析,以5号峰(阿魏酸)为参照峰(见图1),记录指纹图谱(见图2-图4)。
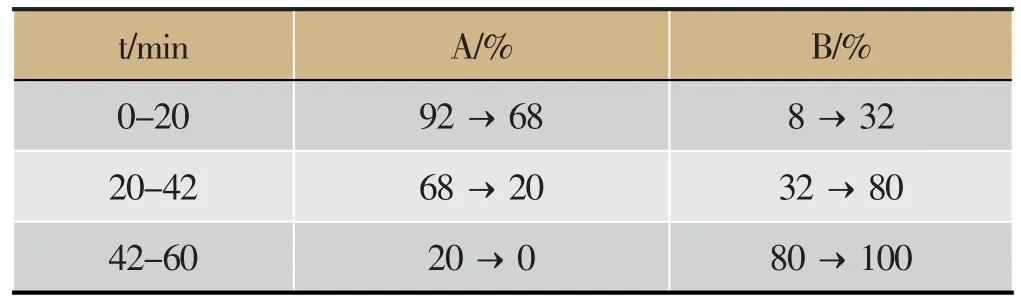
表2 流动相梯度洗脱程序

图1 阿魏酸对照品HPLC图谱

图2 10批当归传统饮片指纹图谱(R为当归传统饮片指纹图谱的共有模式)
2.5.2 共有模式建立及当归传统饮片-破壁粉体-破壁饮片关联性分析
采用“中药色谱指纹图谱相似度评价系统A版”软件,中位数法(时间窗为0.1)分别建立当归传统饮片、破壁粉体及破壁饮片指纹图谱共有模式。结果显示,当归传统饮片、破壁粉体和破壁饮片指纹图谱均标定14个共有峰,其中5号峰为对照峰(阿魏酸)(见图5)。分别计算10批次当归破壁饮片、10批次当归破壁粉体、10批次当归传统饮片的HPLC指纹图谱相似度以及各批次破壁饮片-破壁粉体-传统饮片之间的相似度。结果显示,10批当归传统饮片之间的相似度均大于0.98,当归破壁粉体之间的相似度均大于0.97,当归破壁饮片之间的相似度均大于0.96,各批破壁饮片-破壁粉体-传统饮片之间的相似度均大于0.96(见表3-表7)。

图3 10批当归破壁粉体指纹图谱(R为当归破壁粉体指纹图谱的共有模式)

图4 10批当归破壁饮片指纹图谱(R为当归破壁饮片指纹图谱的共有模式)
由10批破壁饮片的指纹图谱相似度均在0.96
以上可见,不同批次间破壁饮片产品均一性和稳定性较好。由图5和表6、7可见,当归破壁饮片、破壁粉体和传统饮片分别生成的共有模式非常相似,各批次破壁饮片-破壁粉体-传统饮片之间的相关性良好,也表明当归破壁传统饮片制成破壁饮片时,各主要成分均较稳定。

表3 10批次当归传统饮片的相似度

表4 10批次当归破壁粉体的相似度

表5 10批次当归破壁饮片的相似度

表6 10批次当归破壁饮片(UGP)-破壁粉体(UP)- 传统饮片(DP)间的相似度

表7 共有模式相似度结果

图6 当归传统饮片、破壁粉体及破壁饮片的共有模式的比较
2.5.4 当归指纹图谱的主成分分析
采用SIMCA-P 12.0 软件,进行PCA分析,以18个主要色谱峰的峰面积值为变量,得到因子载荷(Loading)图(见图7、图8)和主成分得分图(见图9)。从主成分得分图中可以看出, PC1和PC2共解释了全部变量的98.08%的方差,根据这两个主成分可以概括原始数据的大部分信息内容,基本显示当归破壁饮片-破壁粉体-传统饮片化学成分之间的相似性。区域Ⅰ中即为PC1值适中,PC2值最高的样本,包含当归破壁饮片(1-9)号样本;区域Ⅱ中即为PC1最高,PC2值较低的且离群的样本,包含11、17、30号样本;区域Ⅲ中即为PC1稍低,PC2值最低的样本,包含13、14、15、16、18、19、21、22、23、24、25、26、27、28、29号样本。对因子载荷图进行分析可知,主成分PC1和PC2的载荷量影响最大的是第11个因子(32 min处峰),该因子是影响PC1和PC2主成分的主要因子,将样品分为正负两大区域,当归破壁饮片样品1-10的PC2值为正,当归破壁粉体和传统饮片PC2值为负,使当归破壁饮片-破壁粉体-传统饮片的样品获得了良好的区分,表明在32 min处,当归破壁饮片的化学成分含量远大于当归传统饮片和破壁粉体,也说明利用该主成分分析方法,既能从定性又能从定量角度评价当归破壁饮片-破壁粉体-传统饮片的指纹图谱。

图7 P1主成分因子载荷图

图8 P2主成分因子载荷图

图9 当归样品的指纹图谱的主成分得分图
2.5.3 当归指纹图谱聚类分析
为了更清晰地表达样本与样本之间的差异,笔者对当归传统饮片、当归破壁粉体及破壁饮片各10批的指纹图谱进行聚类分析,取30个样本的平均图谱,基于欧式距离,运用SPSS 16.0对当归的共有峰面积进行分析,得到表征30个样本的当归化学成分的树系图(图10)。30个样本之间欧式距离系数在0-25.0之间变动,当欧式距离系数在0-25间,各测试样品可划分为3个表征群,当归破壁饮片(编号1-9)聚为一类,相关系数>0.96,当归传统饮片(编号21-29)归为一类;当归破壁粉体(12、13、15、18、19)归为一类。除样本16为独立样本,样本10、11、17、30归类错误之外,该聚类分析方法可以有效地区分当归破壁饮片、破壁粉体和传统饮片,可为全面评价当归破壁饮片的质量提供可靠的依据。
2.6 阿魏酸的含量测定
本试验按照《中国药典》2015版中当归的阿魏酸含量测定方法[13],测定当归破壁饮片-破壁粉体-传统饮片阿魏酸,结果见表8。从表中可看出,当归破壁粉体阿魏酸含量均值最高(0.137%),当归破壁饮片次之(0.105%),当归药材低于前二者(0.097%)。
3 讨论
本研究同时对同来源的当归破壁饮片-中间体-传统饮片进行指纹图谱研究并分析三者之间的相关性,结果显示:三者之间全谱相似度均大于0.96,各色谱峰的保留时间几乎一致,但存在一定的含量差异,这表明:当归传统饮片经破壁粉碎并制成破壁饮片的过程中,化学成分组成无明显的变化,破壁技术未造成主成分流失以及新化学成分的转化或生成。

图10 不同当归样品指纹图谱聚类分析结果
在保留时间32 min处,S1破壁粉体相对应的色谱峰远远小于其它9批样品,但S1破壁饮片相对应的色谱峰与其它9批样品较一致,推测破壁粉体作为中间体,因比表面积大,易发生吸潮、氧化等不稳定问题,造成某些不稳定成分含量降低甚至缺失。对该不稳定的成分峰,不适合纳入破壁粉体的共有特征峰。该现象说明,破细胞壁后的粉体制备成破壁饮片后,其稳定性大大提高。在聚类分析和主成分分析中当归破壁饮片均能很好的聚在一起,而当归破壁粉体则出现了聚类错误和孤立样本,这一结果为该推测提供了良好的数据支撑,说明破壁粉体制备成颗粒状破壁饮片的必要性。
本研究测定了当归破壁饮片的原料-中间体-成品的阿魏酸含量,当归破壁粉体阿魏酸含量均值最高,当归破壁饮片次之,当归药材低于前二者,说明经破壁处理,增加了细胞壁内所包裹成分的溶出率,可使其活性成分利用率较传统饮片增加。而破壁粉体的阿魏酸含量虽然最高但却批次间差异较大(RSD>10%),参考有关文献[14,15],阿魏酸见光遇热易分解,在比表面积和吸湿性增大的粉体状态不稳定,导致阿魏酸含量不稳定,进一步提示破壁粉体不适于作为最终成品,破壁饮片制剂工艺稳定,可行。为更全面、科学地评价当归破壁饮片的内在质量,后续仍需在本指纹图谱研究的基础上结合气相色谱和液相色谱等多手段对当归中藁本内酯等挥发油类、黄酮类等重要指标成分的进行含量分析,为当归破壁饮片和传统饮片之间的剂量换算和服用方式提供参考。
运用化学模式识别中药指纹图谱相结合的方法,比较准确地判断当归破壁饮片-破壁粉体-传统饮片样本间的化学成分差异,而破壁粉体既能从定性又能从定量角度评价当归破壁饮片-破壁粉体-传统饮片的指纹图谱,而且反映了指纹图谱的整体性寓于模糊性,为考察和控制破壁饮片质量提供了有效方法。

表8 当归破壁饮片-破壁粉体-传统饮片阿魏酸含量测定结果/%(n=10)
1 冯学花, 梁肖蕾. 当归化学成分与药理作用的研究进展. 广州化工, 2012, 40(22): 16-18.
2 成金乐, 赖智填, 彭丽华. 中药破壁饮片研究. 世界科学技术-中医药现代化, 2014, 16(2): 254-262.
3 梁逸曾, 王兵, 曾茂茂,等. 色谱指纹图谱与中药质量控制. 世界科学技术-中医药现代化, 2010, 12(1): 94-98.
4 容穗华, 林海, 高妮. 不同产地当归中主要有效成分的含量测定.当代医学, 2011, 17(22): 140-141.
5 胡方. 不同产地的当归中紫花前胡素HPLC法含量测定. 中国医药导刊, 2015, 17(5): 533-534.
6 陈建真,吕圭源,宋玉良. 当归头、身、尾的鞣质含量测定. 浙江中医学院学报, 2004, 28(3): 72-76.
7 于淼,李飞,于治国,等. 当归药材中水溶性成分指纹图谱的分析.中国冶金工业医学杂志, 2008, 25(1): 20-22.
8 吴燕燕,尚明英,蔡少青. 当归的化学成分指纹图谱. 药学学报, 2008, 43(7): 728-732.
9 徐魁,韩勇. 当归药材HPLC指纹图谱研究. 中国中医药现代远程教育, 2013, 11(15): 159-161.
10 郭延生,华永丽,杜天玺,等. 基于化学计量学的HPLC指纹图谱在当归炮制品质量控制和识别中的应用. 中国中药杂志, 2010, 35(12): 1551-1555.
11 杨英来,崔方,吴国泰,等.当归药材不同提取部分的指纹图谱分析.中国实验方剂学杂志, 2014, 20(13): 55-60.
12 张亚亚,顾志荣,丁军霞,等.当归不同药用部位近红外漫反射光谱指纹图谱研究.中药材, 2015, 38(7): 1413-1416.
13 国家药典委员会.中华人民共和国药典(一部). 2015年版.北京:中国医药科技出版社, 2015: 133.
14 赵晓燕,杨连威,杨玉芬,等.超微粉碎对当归物理化学特性影响的研究. 世界科学技术-中医药现代化, 2010, 12(3): 418-422.
15 吕海涛,王艳丽,王英芹.当归提取液中阿魏酸稳定性研究.中成药,2008, 30(10): 1555-1557.
Study on the HPLC Fingerprints of the Ultrafine Granular Powder, the Ultrafine Powder and Decoction Pieces Made from Angelicae Sinensis Radix
Liu Xingyun1,2,3, Peng Lihua2,3, Cheng Jinle2,3
(1. Research Center of Chinese Herbal Resource Science and Engineering, Key Laboratory of Chinese Medicinal Resource from Lingnan, Guangzhou University of Chinese Medicine, Guangzhou 510006, China; 2. Key Laboratory of Herbal Ultrafine Granular Powder of Technology and Application, State Administration of Traditional Chinese Medicine, Zhongshan 528437, China; 3. Zhongzhi Pharmaceutical Group Co. Ltd., Zhongshan 528437, China)
This study aimed to establish an effective measurement to exam the quality of three formulations,ultrafine granular powder, ultrafine powder and decoction pieces made from Angelicae sinensis Radix through determining the content of ferulic acid by HPLC fingerprints. RP-HPLC method was performed on an Agilent ZORBAX SB column with a gradient elution using acetonitrile-1% acetic acid at the flow rate of 1 mL·min-1. The column temperature was 30 ℃, and the detective wavelength was 254 nm. In addition, the contents of ferulic acid of the samples were detected according to Chinese Pharmacopoeia (2015 Edition). As a result, the fingerprints of the three formulations of Angelicae sinensis Radix were analyzed. The common pattern of chromatographic fingerprints in which fourteen mutual peaks were marked, was established with 30 batches of samples with 10 batches for each formulation of Angelicae sinensis Radix. The similarity among the fingerprints of the ultrafine granular powder, the granular powder and the decoction pieces were higher than 0.96. The content of ferulic acid in the ultrafine granular powder of Angelicae sinensis Radix was slightly higher than that in its decoction pieces. In conclusion, this method was reliable and stable and with good reproducibility, which was feasible in the area of drug quality control of ultrafine granular powder of Angelicae sinensis Radix.
Angelicae sinensis Radix, ultrafine granular powder, fingerprint, relativity, HPLC
10.11842/wst.2016.09.020
R283.6
A
(责任编辑:马雅静 张志华,责任译审:朱黎婷)
2015-08-06
修回日期:2015-12-11
* 广东省中山市科学技术局资助项目(2008CXY004):丹参等四味中药超微饮片的色谱指纹图谱研究及应用,负责人:成金乐。
** 通讯作者:成金乐,教授,博士生导师,主要研究方向:创新中药研究。
