姜炭超高效液相色谱指纹图谱研究
2016-03-01韩燕全夏伦祝汪永忠沈业兵李钰馨高家荣
韩燕全,洪 燕,夏伦祝,汪永忠,沈业兵,李钰馨,,高家荣
(1.安徽中医药大学第一附属医院 国家中医药管理局中药制剂三级实验室,安徽 合肥 230031;
2.安徽中医药大学中西医结合临床学院,安徽 合肥 230038)
姜炭超高效液相色谱指纹图谱研究
韩燕全1,洪燕2,夏伦祝1,汪永忠1,沈业兵1,李钰馨1,2,高家荣1
(1.安徽中医药大学第一附属医院国家中医药管理局中药制剂三级实验室,安徽 合肥230031;
2.安徽中医药大学中西医结合临床学院,安徽 合肥230038)
[摘要]目的建立姜炭药材的超高效液相色谱-二极管阵列检测器(ultra performance liquid chromatography-photodiode array, UPLC-PDA)指纹图谱。方法采用UPLC BEH色谱柱C18(2.1 mm×100 mm,1.7 μm),流动相为水-乙腈,梯度洗脱;流速0.25 mL/min;柱温30 ℃;检测波长为280 nm;分别对14批样品指纹图谱进行相似度分析、聚类分析和偏最小二乘法判别分析。结果实验指认了指纹图谱15个共有色谱峰中的5个。14批不同产地姜炭药材中9批样品相似度大于0.9,可聚为4大类,其中10个色谱峰的VIP得分值大于1.0。结论所建立的指纹图谱方法简便、稳定、可行,可用于姜炭的质量评价及分类。
[关键词]姜炭;超高效液相色谱指纹图谱;偏最小二乘法判别分析模式识别
中医素有“血见黑则止”的说法,因而炭药作为止血药在中医临床上被广泛采用。炭类中药的质量控制方法是中药学研究的难点,目前除了某些成分的定量测定以外,其控制方法多停留在“炒炭存性”的主观经验判断上,缺少客观、可量化的质量控制手段来全面控制其质量。干姜是姜科植物姜ZingiberofficinaleRosc.干燥根茎;姜炭是由干姜经过清炒而成的炮制品[1]。干姜炮制成炭后,辛味消失,“守而不走”,长于止血温经,主要用于吐血、衄血等脾阳不足病证。现代研究表明,干姜炮制成姜炭后,氨基酸和挥发油等成分已被破坏,姜酚类成分含量降低至微量[2]。2010年版《中华人民共和国药典》对姜炭的质量控制标准是以6-姜辣素(C17H26O4)的含量不得少于0.05%,质控指标单一,有待提高。目前,有关生姜、干姜和炮姜的指纹图谱研究已取得一定进展[3],但尚未见对姜炭指纹图谱的研究。因此,本实验在前期研究基础上,建立了姜炭药材的超高效液相色谱(ultra performance liquid chromatography, UPLC)指纹图谱,并对其进行模式识别分析,以期为全面控制姜炭的质量提供参考依据。
1仪器与试药
Waters UPLC系统主要有四元泵、自动进样器、柱温箱、二极管阵列检测器和Empower 2色谱工作站组成;AG285型十万分之一电子天平:德国赛多利斯公司;KQ-3200B型超声仪:江苏昆山超声仪器有限公司;HFB-200小型中药材粉碎机:湖南省吉首市中州市中药机械厂;DZF-6050型数控真空干燥箱:上海博讯医疗设备厂;色谱纯乙腈:美国SIGMA公司;分析纯试剂甲醇、磷酸等均购自国药集团化学试剂有限公司;蒸馏水为屈臣氏公司生产;0.22 μm微孔滤膜购自北京八方世纪科技有限公司。
实验用5种姜酚类对照品的批号和来源:姜酮(批号 122-485)、6-姜酚(批号 23513-146)、8-姜酚(批号 23513-088)、10-姜酚(批号 23513-157)对照品均购自上海鼎瑞化工有限公司,;6-姜烯酚(批号 12121803)对照品购自四川省成都曼斯特公司,5种对照品含量均大于98.0%,并经1H-NMR、13C-NMR、MS、IR和UV等光谱检测确认其结构。14份市售干姜药材购自全国不同地区药店,经安徽中医药大学第一附属医院高家荣主任中药师鉴定均为姜科植物姜ZingiberofficinaliesRose.的干燥根茎。姜炭炮制方法采用《中华人民共和国药典》2010年版附录ⅡD项下炒炭法,主要过程为取干姜块,炒至表面黑色、内部棕褐色,即得到姜炭药材,样品信息见表1。
2方法与结果
2.1色谱条件色谱柱为Waters Acquity UPLC BEH C18柱(2.1 mm×100 mm,1.7 μm);流动相为乙腈(A)-水溶液(B),梯度洗脱程序为:0~10 min,25%~75% A;10~15 min,75% A;15~16.5 min, 75%~100% A;16.5~18 min, 100% A; 18~20 min,100%~25% A;流速为0.25 mL/min;柱温为30 ℃;检测波长为280 nm;样品进样量为2 μL。
2.2对照品溶液的制备精密称取姜酮5.6 mg、6-姜酚5.0 mg、8-姜酚6.0 mg、6-姜烯酚5.8 mg和10-姜酚6.6 mg,各对照品分别置于容量瓶中,加甲醇定容,制备得到对照品母液;再分别吸取5种对照品母液一定容积置于同一10 mL容量瓶并定容,得终浓度分别为姜酮28 μg/mL、6-姜酚25 μg/mL、8-姜酚30 μg/mL、6-姜烯酚58 μg/mL和10-姜酚33 μg/mL的5种对照品混合溶液,冷藏备用。
2.3供试品溶液的制备取表1中各姜炭样品,粉碎,过40目筛;精密称取姜炭粉末0.5 g,至10 mL棕色量瓶中,加入90%乙醇至刻度,150 W,40 Hz超声提取40 min,放冷,再加入90%乙醇补足损失量,摇匀,再用0.22 μm微孔滤膜过滤,留取续滤液,供指纹图谱分析用。
2.4方法学考察
2.4.1精密度试验取S6号(四川产)姜炭供试品溶液,按照“2.1”项下的色谱条件,自动进样器连续进样6次,记录指纹图谱,对15个共有色谱峰的相对保留时间和相对峰面积的RSD计算结果表明,相对保留时间的RSD为0.02%~0.57%,相对峰面积的RSD为1.14%~2.72%;采用《中药色谱指纹图谱相似度评价系统软件》(2004版A)对6个样品的指纹图谱进行相似度评价,结果6次进样指纹图谱的相似度≥0.992,表明仪器精密度较好,可用于指纹图谱测定。
2.4.2重复性试验取S5号(四川产)姜炭药材粉末6份,每份0.5 g左右,按照“2.3”项下方法制备供试品溶液并测定其指纹图谱,并计算15个共有色谱峰的相对保留时间和相对峰面积的RSD。结果表明,相对保留时间的RSD为0.28%~0.52%,相对峰面积的RSD为0.65%~2.87%,以《中药色谱指纹图谱相似度评价系统软件》(2004版A)评价,6份样品的相似度≥0.981,表明样品处理方法的重复性较好。
2.4.3稳定性试验取S2号(山东产)姜炭供试品溶液,按照“2.1”项下的色谱条件,分别在样品处理完成的第0、4、8、12、16、24 h进样,记录色谱图。计算15个共有色谱峰的相对保留时间和相对峰面积的RSD。结果表明,相对保留时间的RSD为0.13%~0.57%,相对峰面积的RSD为1.53%~2.16%,以《中药色谱指纹图谱相似度评价系统软件》(2004版A)评价,6个时间点的指纹图谱相似度≥0.951,表明样品溶液至少在24 h内较稳定。
2.4.4数据处理相似度评价采用国家药典委员会《中药色谱指纹图谱相似度评价系统软件》(2004A版);采用SPSS 16.0分析软件进行系统聚类分析;偏最小二乘法判别分析(partial least squares-discriminate analysis, PLA-DA)采用SIMCA 13.0分析软件进行。
2.5指纹图谱测定与建立[4-5]分别取“2.2”项下的混合对照品溶液和14批姜炭供试品溶液,按“2.1”项下色谱条件进行测定,分别得到5种对照品混合溶液和14批不同产地姜炭样品的指纹图谱。采用《中药色谱指纹图谱相似度评价系统软件》(2004版A)处理所得到的姜炭药材图谱,并生成姜炭药材共有模式的对照指纹图谱,5种对照品混合液和样品的共有模式色谱图见图1。
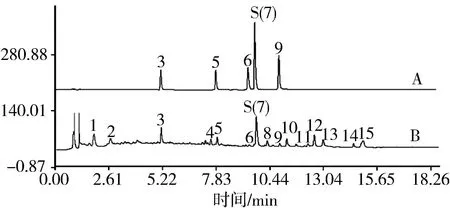
图1 混合对照品(A)和姜炭药材共有模式(B)色谱图
《中药色谱指纹图谱相似度评价系统软件》(2004版A)共匹配了15个色谱峰。通过与对照品保留时间比对,共指认15个色谱峰中的5个峰,分别是3号峰(姜酮),5号峰(6-姜酚),6号峰(8-姜酚),7号峰(6-姜烯酚),9号峰(10-姜酚)。其中7号色谱峰(6-姜烯酚)出峰时间位于中间,面积最大且分离度较好,设为参照色谱峰。
2.6相似度评价将实验所得的14批姜炭药材UPLC图谱以分析仪器关联(analytical instrument association, AIA)格式导出,再依次导入《中药色谱指纹图谱相似度评价系统软件》(2004版A)软件,以S1号(河北产)样品图谱作为参照图谱进行指纹匹配,14批姜炭样品匹配了15个共有色谱峰,并建立了姜炭药材的共有模式指纹图谱。14批姜炭样品的相似度计算结果和叠加指纹图谱分别见表1和图2。结果显示,14批药材中相似度低于0.9的有5批,其中4批产地为四川。由于采用的炮制工艺较为均一,推测姜炭药材的质量差异主要发生在药材的生长、干燥、储存等环节。
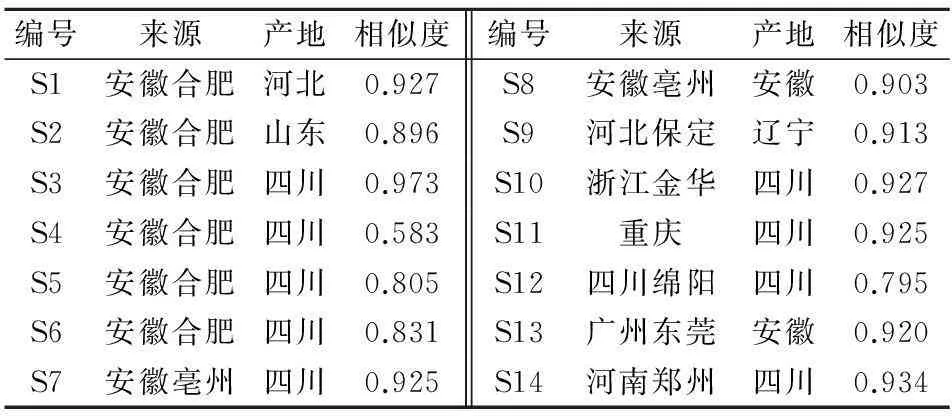
表1 14份姜炭样品来源
2.7系统聚类分析以共有色谱峰峰面积的积分值作为数据源,采用SPSS 16.0软件对14批姜炭样品进行系统聚类分析,采用组间平均数联结法(average linkage between groups),以夹角余弦作为样品相似度的距离公式,聚类树形图见图3。根据图3聚类分析结果,14批姜炭样品大致可分为聚为4大类,Ⅰ类包括S1和S14,质量最好;Ⅱ类包括S7、S11、S9、S10、S13、S8、S3、S2和S5,质量较好;Ⅲ类包括S6和S12,质量次之;S4单独成类,与前3类样品差异较大,质量最差。各样品聚类分析的相关性与指纹图谱相似度之间的相关性,总体趋势较一致。

图2 14批姜炭药材UPLC指纹图谱叠加图
2.8PLS-DA[6-7]将14批姜炭的共有样品色谱峰面积数据导入SIMCA 13.0软件,进行PLS-DA分析。图4(A)可见14批样品均在95%置信区间内,
分布在t1轴的左右两侧,左侧有3批样品,右侧有11批样品,根据相似度和聚类分析结果可见,左侧的3批样品即S12、S6、S4相似度较低,与其他样品聚类距离较远,属质量较差样品;右侧的11批姜炭又被t2轴分为上下2类,上面的4个样品属质量较佳的样品,下面7批样品属质量居中,应属合格样品,相似度、聚类分析和PLS-DA的分析结果基本一致,可相互印证。PLS-DA分析各色谱峰(F)的VIP得分图表明,色谱峰变量对样品分类的影响大小排序为F15>F3>F14>F9>F7>F12>F13>F8>F10>F5>F6>F2>F4>F11>F1;其中VIP值>1.0的色谱峰共有10个,分别是F15、F3、F14、F9、F7、F12、F13、F8、F10、F5,说明姜炭的质量差异是由总体色谱峰群的影响所导致的。

图3 姜炭样品聚类分析结果

图414批姜炭样品PLS-DA图(A.样品得分图;B.各色谱峰变量投影重要性分析得分图)
4讨论
姜既是必不可少的调味品,又是临床常用的中药材,其常用炮制品有干姜、炮姜和姜炭等,其中姜炭是由干姜高温炒炭存性的炮制品。姜中含多种姜酚类活性成分,具有抗血小板聚集,抗氧化和抗炎、降糖和抗肿瘤等多种作用[8-9]。而干姜炒炭后辛味消失,其温中作用弱于干姜和炮姜,更擅长温经止血,可用于各种虚寒性出血。姜酚类成分具有多酚羟基结构特点,在酸、碱、热等环境下多酚羟基结构易脱水形成姜烯酚[10],这可能是干姜制炭后辛味消失,具有温经止血作用的机制之一。
炭药炮制工艺的关键是“炒炭存性”,即既要保证药物在高温下其外部色泽炭化变黑,又要保持药物内部固有的色、嗅、味性能,切忌灰化[11]。中药制成炭药后多具有止血作用,这是制炭后药材多成分变化共同的结果,因而在炭药的质量控制方面,单靠测定已知的单一指标成分的含量是不够的,应从指纹图谱等角度为炮制工艺及质量控制提供更全面的参考。色谱指纹图谱鉴别方法可以较全面地反映中药化学成分的种类与数量,进而反映中药的质量和中医用药所体现的整体疗效[12]。
本实验过程中对样品处理方法、流动相系统、柱温、流速、检测波长和进样量等试验条件均进行了优化,最终得到最佳的实验条件,建立了姜炭药材的指纹图谱。通过相似度分析、聚类分析和PLS-DA模式识别分析相互印证,能将各产地药材质量较清晰地区别和分类。本次样品多购自市售药材市场,但姜炭为姜的炮制品,受产地、采收年限和炮制过程的影响较大,实验结果基本反映了目前市售药材的质量状况。实验所建立的指纹图谱对于控制姜炭的药材质量有一定的参考意义,结合实际情况对于姜炭的指纹图谱质量控制,可以考虑以相似度在0.9以上的样品为参照。
参考文献:
[1]国家药典委员会.中华人民共和国药典:一部[M].北京:中国医药科技出版社,2010:13.
[2]石宇华.干姜的质量标准及干姜、炮姜和姜炭的化学成分比较研究[D].成都:成都中医药大学,2008:30-51.
[3]王维皓,李娟,高慧敏,等.从HPLC特征图谱分析姜在炮制过程中的化学成分变化[J].药物分析杂志,2009,29(8):1248-1252.
[4]刘芷,贾英,赵旭,等.五味子的UPLC指纹图谱研究[J].中草药,2014,45(11):1631-1633.
[5]唐峥,毕开顺,韩飞,等.栀子大黄汤UFLC指纹图谱研究[J].中草药,2014,45(3):367-372.
[6]云琦,刘青旺,马小华,等.新疆桃仁HPLC-UV指纹图谱研究及在地区分类中的应用[J].中国中药杂志,2014,39(5):860-866.
[7]周喜丹,唐力英,周国洪,等.HPLC指纹图谱鉴别葶苈子与车前子[J].中国中药杂志,2015,40(2):275-279.
[8]Rahmani AH, Shabrmi FM, Aly SM. Active ingredients of ginger as potential candidates in the prevention and treatment of diseases via modulation of biological activities[J]. Int J Physiol Pathophysiol Pharmacol,2014,6(2):125-136.
[9]Ghlissi Z, Atheymen R, Boujbiha MA, et al. Antioxidant and androgenic effects of dietary ginger on reproductive function of male diabetic rats[J]. Int J Food Sci Nutr, 2013,64(8):974-978.
[10]黄雪松,宴日安,吴建中.姜酚的生物活性述评[J].暨南大学学报(自然科学版),2005,26(3):434-439.
[11]徐慧青.浅析炭药的炮制质量控制[J].浙江医学教育,2009,8(4):43-44.
[12]罗军,卿娟,张丽艳,等.续断生品与酒炙品HPLC指纹图谱及其成分差异分析[J].中药材,2015,38(3):493-496.
A Study of Ultra-Performance Liquid Chromatography Fingerprint of Ginger Charcoal
HANYan-quan1,HONGYan2,XIALun-zhu1,WANGYong-zhong1,SHENYe-bing1,LIYu-xin1,2,GAOJia-rong1
(1.TheFirstAffiliatedHospitalofAnhuiUniversityofChineseMedicine&Grade3LaboratoryofTraditionalChineseMedicinePreparation,StateAdministrationofTraditionalChineseMedicine,AnhuiHefei230031,China; 2.ClinicalSchoolofTraditionalChineseandWesternMedicine,AnhuiUniversityofChineseMedicine,AnhuiHefei230038,China)
[Abstract]ObjectiveTo establish the fingerprint of ginger charcoal by ultra-performance liquid chromatography-photodiode array (UPLC-PDA). MethodsThe UPLC BEH column C18(2.1 mm × 100 mm, 1.7 μm) was used with a mobile phase of water-acetonitrile for gradient elution at a flow rate of 0.25 mL/min, a column temperature of 30 ℃, and a detection wavelength of 280 nm; similarity analysis, cluster analysis, and partial least squares discriminant analysis were performed for the fingerprints of 14 batches of samples. ResultsThe laboratory test identified 5 out of 15 common chromatographic peaks in the fingerprints; of all the 14 batches of ginger charcoal produced in different places, 9 had a similarity higher than 0.9 and were clustered into 4 categories, and 10 chromatographic peaks had a VIP score higher than 1.0. ConclusionThe method to establish fingerprints is simple, stable, and feasible, and can be applied for the quality evaluation and categorization of ginger charcoal.
[Key words]Ginger charcoal; UPLC fingerprint; Partial least squares discriminant analysis for pattern recognition
收稿日期:(2015-07-27;编辑:姚实林)
通信作者:高家荣,zyfygjr2006@163.com
作者简介:韩燕全(1978-),男,博士,副主任中药师
基金项目:国家自然科学基金项目(81102811);安徽省自然科学基金项目(10040606Q40);国家中医药重点学科临床中药学建设项目(国中医药人教发[2012]32号)
[中图分类号]R284.1[DOI]10.3969/j.issn.2095-7246.2016.01.025
