福司氟康唑杂质——2-(2,4-二氟苯基)-1-(1H-1,2,4-三氮唑-1-基)-3-(4H-1,2,4-三氮唑-4-基)-2-丙基磷酸二氢酯的合成
2016-03-01章有国郑仁林袁小红李良春
章有国, 李 强, 黄 毅, 郑仁林, 袁小红, 李良春
(西南科技大学 生命科学与工程学院,四川 绵阳 621010)
·制药技术·
福司氟康唑杂质——2-(2,4-二氟苯基)-1-(1H-1,2,4-三氮唑
-1-基)-3-(4H-1,2,4-三氮唑-4-基)-2-丙基磷酸二氢酯的合成
章有国, 李强, 黄毅, 郑仁林, 袁小红, 李良春*
(西南科技大学 生命科学与工程学院,四川 绵阳621010)
摘要:以1,3-二氟苯为起始原料,依次经傅-克酰基化,1H-三氮唑取代,环氧化,胺解,4H-三氮唑环化,磷酸酯化和钯碳加氢反应等7步反应合成了福司氟康唑的主要杂质——2-(2,4-二氟苯基-1-(1H-1,2,4-三氮唑l-1-基)-3-(4H-1,2,4-三氮唑-4-基)-2-丙基磷酸二氢酯,纯度98%,总收率7.8%,其结构经1H NMR确证。
关键词:福司氟康唑; 杂质; 4H-1,2,4-三氮唑-4-基; 药物合成
Synthesis of Impurity of Fosfluconazole——2-(2,4-
通信联系人: 李良春,副研究员, E-mail: lilc76@gmail.com
福司氟康唑(Fosfluconazole, Ⅰ),化学名为2,4-二氟-α,α-二(1H-1,2,4-三唑-1-基甲基)苯甲醇磷酸二氢酯,是辉瑞公司生产的第三代唑类抗深部真菌药物氟康唑的前体药物[1]。Ⅰ可以以盐形式电离,使其水溶解度显著增加,并在人体内碱性磷酸酯酶作用下裂解,从而释放出活性药物氟康唑达到治疗目的。Ⅰ具有作用迅速、刺激性小、副作用小和安全性较高等优点[2]。
合成Ⅰ的过程中会有一些副产物生成,如2-(2,4-二氟苯基)-1-(1H-1,2,4- 三氮唑-1-基)-3-(4H-1,2,4-三氮唑-4-基)-2-丙基磷酸二氢酯(1),氟康唑(2)和2-(2-氟苯基)-α,α-(1H-1,2,4-三氮唑-1-基) 2-丙基磷酸二氢酯(3)等[3](Chart 1)。
为提高Ⅰ的用药安全性并控制其质量,需对Ⅰ中的杂质进行深入研究。1的结构虽已比较清楚,但其合成方法尚无文献报道[4-14]。本文以间二氟苯(4)为起始原料,经傅克酰基化[5-6],1H-三氮唑取代[8],环氧化[7,15],胺解[16],4H-三氮唑环化[16-17],磷酸酯化和钯碳加氢反应[18-19]等7步反应合成了Ⅰ的主要杂质1(Scheme 1),纯度98%,总收率7.8%,其结构经1H NMR确证。
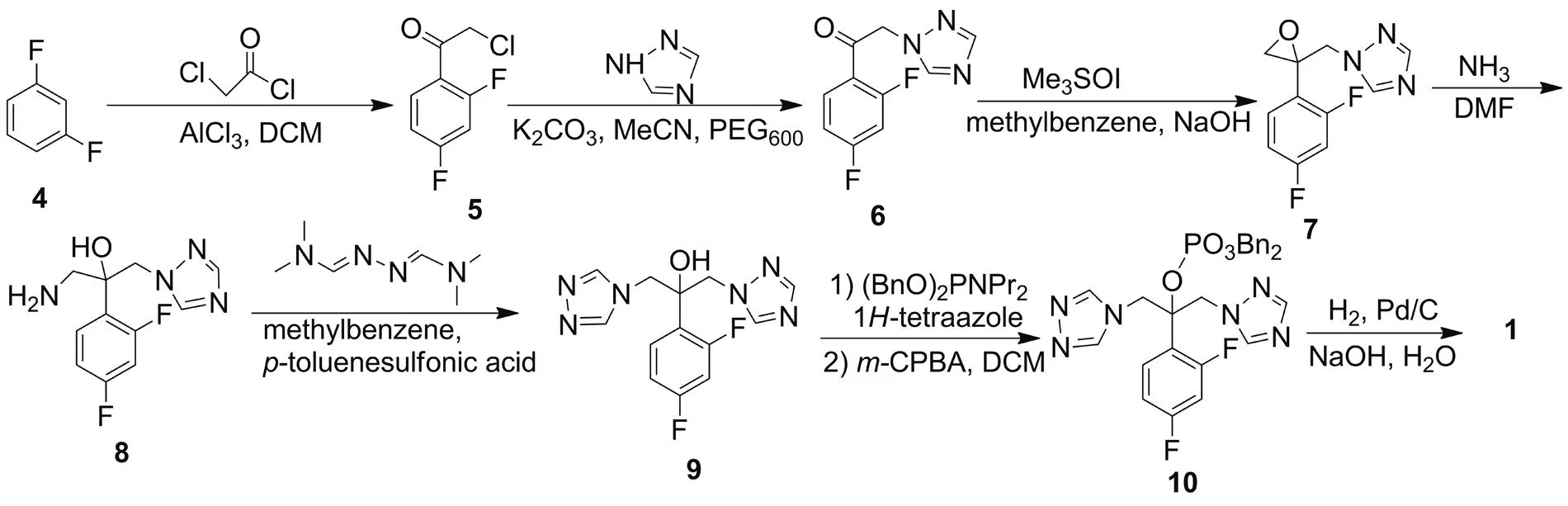
Scheme 1
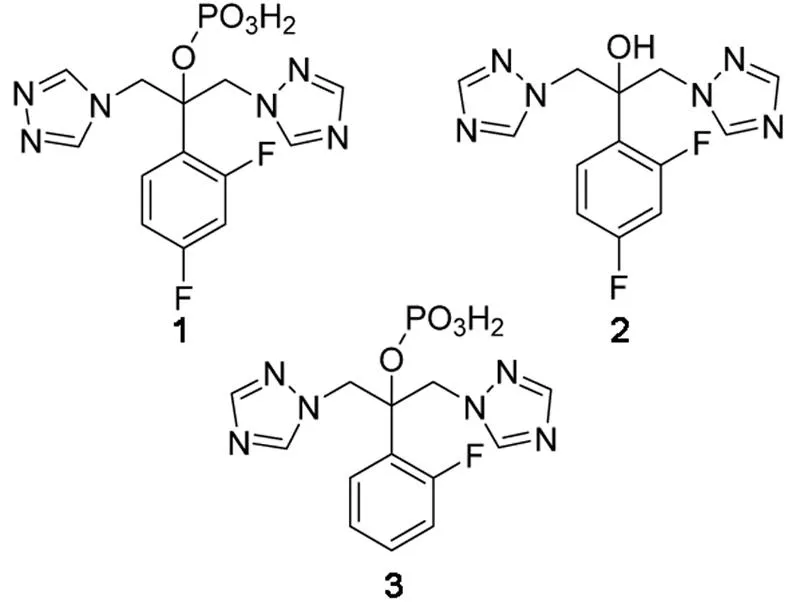
Chart 1 Ⅰ的主要杂质
1实验部分
1.1仪器与试剂
Bruker Avance 600 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Aglient Explise XDB型高效液相色谱仪[C18柱:4.5 mm×150 mm,流动相:20 mmol·L-1磷酸二氢钾溶液/乙腈(V/V=55/45),检测波长:260 nm;流速:1.0 mL·min-1,柱温25 ℃,进样量为20 μL]。
所用试剂均为化学纯或分析纯。
1.2合成
(1)α-氯-2,4-二氟苯乙酮(5)的合成
在单口瓶中加入AlCl340 g和4 19.8 mL(0.2 mol),密封通氮,搅拌下缓慢滴加氯乙酰氯18 mL(0.2 mol),滴毕,于常温反应6 h。反应液倒入冰水中,用二氯甲烷萃取,合并有机相,用无水硫酸钠干燥;过滤,滤液减压浓缩得淡黄色固体5 36 g,收率95%;1H NMRδ: 8.04(td,J=8.5 Hz, 6.6 Hz, 1H), 7.09~6.99(m, 1H), 6.93(ddd,J=11.0 Hz, 8.5 Hz, 2.3 Hz, 1H), 4.71(d,J=3.0 Hz, 1H)。
(2)α-(1H-1,2,4-三氮唑-1-基)-2,4-二氟苯乙酮的合成(6)[20]
在单口瓶中加入K2CO34.26 g和1H-1,2,4-三氮唑4.83 g(70 mmol),搅拌下加入乙腈50 mL和5 9.5 g(50 mmol),回流反应5 h。滤除K2CO3,滤液减压浓缩,残余物用乙酸乙酯溶解,用无水硫酸钠干燥;减压浓缩后经硅胶柱层析[洗脱剂:V(EtOAc) ∶V(Et2O)=1 ∶2]纯化得淡黄色固体6 6.87 g,收率60%;1H NMRδ: 8.21(s, 1H), 8.05(td,J=8.6 Hz, 6.5 Hz, 1H), 8.01(s, 1H), 7.06(ddd,J=9.2 Hz, 7.6 Hz, 2.4 Hz, 1H), 6.98(ddd,J=11.0 Hz, 8.4 Hz, 2.4 Hz, 1H), 5.59(d,J=3.5 Hz, 1H)。
(3) 1-[α-(2,4-二氟苯基)]-2,3-环氧丙基-1H-1,2,4-三氮唑(7)的合成[15,20]
在单口瓶中加入三甲基碘化亚砜3.30 g和新制20%氢氧化钾溶液10 mL,于室温搅拌2 h。加入6 2.23 g(10 mmol)和甲苯80 mL,于50 ℃反应2 h(TLC检测)。冷却至室温,分液,水层用二氯甲烷萃取,合并有机相,用无水硫酸钠干燥;减压浓缩后经硅胶柱层析[洗脱剂A:V(DCM) ∶V(MeOH)=20 ∶1]纯化得无色油状液体7 1.65 g,收率69%;1H NMRδ: 8.07(s, 1H), 7.87(s, 1H), 7.19(dd,J=14.9 Hz, 8.3 Hz, 1H), 6.89~6.76(m, 1H), 4.88~4.43(m, 1H), 2.92(dd,J=25.7 Hz, 4.7 Hz, 1H)。
(4) 1-氨基-2-(2,4-二氟苯基)-3-(1H-1,2,4-三氮唑-1-基)-2-丙醇(8)的合成[15]
在单口瓶中加入7 2.32 g(10 mmol), DMF 40 mL和氨水20 mL,搅拌下回流(80 ℃)反应4 h。旋蒸除溶,残余物经硅胶柱层析(洗脱剂:A)纯化得黄色固体8 1.9 g,收率75%;1H NMRδ: 8.08(s, 1H), 7.82(s, 1H), 7.56(td,J=8.9 Hz, 6.7 Hz, 1H), 6.81(tdd,J=14.1 Hz, 8.4 Hz, 2.4 Hz, 1H), 4.59(d,J=2.8 Hz, 1H), 3.20(dd,J=13.0 Hz, 0.9 Hz, 1H), 2.98(dd,J=13.0 Hz, 1.2 Hz, 1H)。
(5) 2-(2,4-二氟苯基)-1-(1H-1,2,4-三氮唑-1-基)-3-(4H-1,2,4-三氮唑-4-基)-2-丙醇(9)的合成[4]
在双口瓶中加入8 252 mg(1 mmol),N,N-二甲基甲酰肼284 mg(2 mmol)和一水对甲苯磺酸38 mg,氮气保护下加入甲苯5 mL,搅拌下回流(110 ℃)反应24 h。冷却至室温,析出固体;过滤,滤饼用二氯甲烷洗涤,干燥得白色固体9 243 mg,收率79%;1H NMRδ: 8.32(s, 1H), 8.19(s, 2H), 7.83(s, 1H), 7.24(ddd,J=11.8 Hz, 9.1 Hz, 2.6 Hz, 1H), 7.15(td,J=9.0 Hz, 6.7 Hz, 1H), 6.91(td,J=8.4 Hz, 2.6 Hz, 1H), 4.72(d,J=14.5 Hz, 1H), 4.59(d,J=14.7 Hz, 1H), 4.54(d,J=14.5 Hz, 1H), 4.39(d,J=14.6 Hz, 1H)。
(6) 2-(2,4-二氟苯基)-1-(1H-1,2,4-三氮唑-1-基)-3-(4H-1,2,4-三氮唑-4-基)-2-丙基磷酸二苄基酯(10)的合成
在单口瓶中加入9 306 mg(1 mmol), 1H-四氮唑210 mg(3 mmol)和二苄基二异丙基氨基磷酸酯690 mg(2 mmol),氮气保护,搅拌下于室温加入DCM 5 mL,反应2 h。冷却至0 ℃,滴加溶有间氯过氧苯甲酸(m-CPBA)242 mg的DCM(2 mL)溶液,滴毕,搅拌下于室温反应1 h。有机相依次用饱和亚硫酸溶液和饱和碳酸钠溶液洗涤,无水硫酸钠干燥,减压浓缩后经硅胶柱层析(洗脱剂:A=30 ∶1)纯化得白色固体10 301 mg,收率54%;1H NMRδ: 8.21(s, 1H), 8.18(s, 2H), 7.82(s, 1H), 7.36(td,J=4.6 Hz, 1.7 Hz, 5H), 7.28~7.26(m, 3H), 7.24(dd,J=6.4 Hz, 3.1 Hz, 2H), 6.74(tdd,J=12.1 Hz, 7.8 Hz, 4.5 Hz, 2H), 6.64~6.59(m, 1H), 4.99(d,J=2.9 Hz, 1H), 4.97(d,J=2.5 Hz, 1H), 4.95(s, 2H), 4.92(dd,J=9.5 Hz, 4.7 Hz, 2H), 4.90(s, 1H)。
(7) 1的合成
搅拌下,在反应釜中加入10 3 g(5 mmol),纯水20 mL, NaOH 0.4 g和10%Pd/C 0.3 g,密封,用H2置换4次,加压至0.4 MPa,于室温反应36 h。滤除钯碳,滤液用浓盐酸调至pH 1~2,搅拌30 min;析出固体,过滤,滤饼用丙酮(10 mL)洗涤,干燥得白色固体1 1.237 g,收率62%;1H NMRδ: 8.53(s, 1H), 8.37(s, 1H), 7.76(s, 1H), 7.21(ddd,J=11.8 Hz, 9.0 Hz, 2.4 Hz, 1H), 7.08(td,J=9.0 Hz, 6.8 Hz, 1H), 6.86(td,J=8.5 Hz, 2.5 Hz, 1H), 5.30(d,J=15.1 Hz, 1H), 5.15(d,J=15.2 Hz, 1H), 4.97(dd,J=19.2 Hz, 15.3 Hz, 1H)。
2结果与讨论
2.1合成
对1进行逆合成分析发现,1可由氟康唑中含有的杂质9经磷酸酯化、钯碳加氢等反应合成[4]。因此,如果我们能通过改变合成条件,使9由副产物变为主产物,合成过程将被大大简化。文献[4]指出,在不同反应条件下,1H-1,2,4-三氮唑的1-位和4-位取代产物比例不同。如碱性条件下,生成4-取代三氮唑[21]。但实际反应中并未能制得9。故利用1H-1,2,4-三氮唑直接反应并不能合成我们的预期产物。后改用直接成环的方法才合成4H-1,2,4-三氮唑产物。根据文献[17]方法,利用7与氨水回流反应生成8,再使其成环。实验过程中,我们发现8与甲酰肼在乙醇、甲醇中加热反应,或与二甲酰肼在吡啶中加入三甲基氯硅烷加热至110 ℃反应,均未能合成9。最后,我们以对甲苯磺酸为催化剂,8与N,N-二甲基甲酰肼在甲苯中反应,合成了关键中间体9。
2.2纯度

Time/min
图1为1的HPLC谱图。由图1可见,1纯度98%,符合药典要求。
3结论
合成了福司氟康唑的主要杂质——2-(2,4-二氟苯基-1-(1H-1,2,4-三氮唑l-1-基)-3-(4H-1,2,4-三氮唑-4-基)-2-丙基磷酸二氢酯(1),纯度较高(98%)。1的合成解决了杂质对照品的来源问题,对Ⅰ的质量控制具有一定意义。
参考文献
[1]Hecker S J, Erion M D. Prodrugs of phosphates and phosphonates[J].J Med Chem,2008,51(8):2328-2245.
[2]国大亮,孙晋瑞,刘振国. 福司氟康唑[J].齐鲁药事,2005,24(1):60-60.
[3]王琰,王明娟,胡昌勤. 磷氟康唑有关物质分析方法的筛选与优化[J].药物分析质,2010,30(5):883-886.
[4]莫安国,谢庆潮,吴瑞芳,等. 氟康唑相关物质的分离与鉴定[J].中国医药工业杂志,1995,26(1):18-19.
[5]Richardson K. Antifungal 1,3-bis-triazolyl-2-propanol derivative:US 4 554 286[P].1985.
[6]吴春丽,李幸,施秀芳,等. 氟康唑的合成工艺研究及优化[J].中国药物化学杂志,2011,21(4):304.
[7]盛春泉,陈宜,张万年,等. 正交设计法优化氟康唑关键中间体的合成工艺[J].中国药物化学杂志,2002,12(6):344-346.
[8]Xu Y, Sheng C, Wang W,etal. Structure-based rational design,synthesis and antifungal activity of oxime-containing azole derivatives[J].Bioorg Med Chem Lett,2010,20(9):2942-2945.
[9]Sobue S, Tan K, Layton G,etal. The effects of renal impairment on the pharmacokinetics and safety of fosfluconazole and fluconazole following a single intravenous bolus injection of fosfluconazole [J].Brit J Clin Pharm,2004,57(6):773-784.
[10]Potts K. The chemistry of 1,2,4-triazoles[J].Chem Rev,1961,61(2):87-127.
[11]Osenderichina, Veinberga. Processes for the preparation of 1,3-bis(1,2,4-triazol-1-yl)-propan-2-ol derivatives:US 5 484 936[P].1996.
[12]Boucher H W, Groll A H, Chiou C C,etal. Newer systemic antifungal agents[J].Drugs,2004,64(18):1997-2020.
[13]Sajiki H, Hirota K. A novel type of Pd/C-catalyzed hydrogenation using a catalyst poison:Chemoselective inhibition of the hydrogenolysis forO-benzyl protective group by the addition of a nitrogen-containing base[J].Tetrahedron,1998,54(46):13981-13996.
[14]Bentley A, Butters M, Green S P,etal. The discovery and process development of a commercial route to the water soluble prodrug, fosfluconazole[J].Org Proc Res Devel,2002,6(2):109-112.
[15]Borate H B, Sawargave S P, Maujan S R. A short synthesis of 3,6-disubstitutedN-2-thienyl/aryl-indoles[J].Tetrahedron Lett,2009,50(47):6562-6566.
[16]Willam J H, Robert J S. Matalloenzyme inhibitor compounds:US 201 203 298 02A[P].2012.
[17]Bartlett R K , Humphrey I R. Transaminations ofN,N-dimethylformamide azine [J].J Chem Soc,1967,1664-1666.
[18]C W 默蒂肖, S 格林, P T 斯特菲森. 用于治疗的三唑衍生物:CN 10 852 13C[P].2002.
[19]冀亚飞,忻许,宗志敏,等. 钯/碳催化3,4,5-三甲氧基苯甲醛还原反应[J].J Chem Indust Eng,2002,52(2):207-211.
[20]Lebouvier N, Giraud F, Corbin T,etal. Efficient microwave-assisted synthesis of 1-(1H-indol-1-yl)-2-phenyl-3-(1H-1,2,4-triazol-1-yl)propan-2-ols as antifungal agents[J].Tetrahedron Lett,2006,47(36):6479-6483.
[21]Olofoukendall R V J. Protection by acylation in the selective alkylation of heterocycles[J].J Org Chem,1970,35(7):2246-2248.
Difluorophenyl)-1-(1H-1,2,4-triazol-1-yl)-3-(4H-
1,2,4-triazol-4-yl)propan-2-yl Dihydrogen Phosphate
ZHANG You-guo,LI Qiang,HUANG Yi,
ZHENG Ren-lin,YUAN Xiao-hong,LI Liang-chun*
(School of Life Science and Engineering, Southwest University of Science and Technology, Mianyang 621010, China)
Abstract:The main impurity of Fosfluconazole, 2-(2,4-difluoro-Phenyl)-1-(1H-1,2,4-triazol-1-yl)-3-(4H-1,2,4-triazol-4-yl)propan-2-yl dihydrogen phosphateoxalate with purity of 98% and total yield of 7.8%, was synthesized by a seven-step reaction of Friedel-Crafts reaction, nucleophilic substitution, Corey-Chaykovsky reaction, phosphorylation and catalytic hydrogenation, using 1,3-difluorobenzene as starting material. The structure was confirmed by1H NMR.
Keywords:Fosfluconazole; impurity; 4H-1,2,4-triazole; drug synthesis
作者简介:章有国(1988-),男,汉族,四川绵阳人,硕士研究生,主要从事有机合成的研究。
基金项目:国家自然科学基金资助项目(21002099); 西南科技大学杰出青年基金资助项目(13zx9104)
收稿日期:2015-11-04;
修订日期:2015-12-21;
中图分类号:R914.5; O626.26
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.02.15370
