新型O6-苄基鸟嘌呤衍生物的合成及其细胞毒性
2016-03-01李思思彭瑞增
李思思, 彭瑞增
(1. 中国疾病预防控制中心 环境与健康相关产品安全所,北京 100021;
2. 北京工业大学 生命科学与生物工程学院,北京 100124)
·研究简报·
新型O6-苄基鸟嘌呤衍生物的合成及其细胞毒性
李思思1*, 彭瑞增2
(1. 中国疾病预防控制中心 环境与健康相关产品安全所,北京100021;
2. 北京工业大学 生命科学与生物工程学院,北京100124)
摘要:以O6-苄基鸟嘌呤(O6-BG)为起始原料,经4步反应合成了一个新型的O6-BG衍生物——4-硝基苄基-[6-(苄氧基)-9H-嘌呤-2]氨基甲酸酯(4),其结构经1H NMR和HR-ESI-MS表征。用CCK-8法研究了4对人脑神经胶质细胞(SF126, SF763和SF767)的细胞毒性。结果表明:在低氧环境下,4与ACUN的协同作用对SF126, SF763和SF767均有较好的抑制活性,其IC50分别为0.04 mM, 0.1 mM和0.03 mM,优于阳性对照药O6-BG。
关键词:O6-苄基鸟嘌呤; 低氧激活; 4-硝基苄基-[6-(苄氧基)-9H-嘌呤-2]氨基甲酸酯; 合成; 细胞毒性
氯乙基亚硝基脲类化合物(CENUs)是治疗神经胶质瘤等恶性肿瘤的重要化疗药物。该药物通过产生DNA股间交联[1-2],抑制细胞正常复制,从而发挥抗癌作用。O6-烷基鸟嘌呤-DNA烷基转移酶(AGT)是存在于细胞内的DNA修复蛋白,可修复CENUs对肿瘤细胞DNA产生的烷化损伤[3-4],导致肿瘤细胞对化疗药物产生耐药性[5]。近年来,有多种AGT抑制剂与烷化剂联合应用于肿瘤患者,其中O6-苄基鸟嘌呤(O6-BG)已进入临床实验阶段。但O6-BG不具有靶向性,在肿瘤细胞内发挥增敏作用的同时,亦会抑制正常细胞内AGT活性,增加药物敏感性,导致烷化剂的使用剂量大量降低至无效水平之下[6-8]。因此,合成具有靶向性的AGT抑制剂对提高CENUs抗癌效果具有较大的临床意义。
药物作用环境对药效有较大影响。文献[9-13]研究表明,实体肿瘤中会出现缺氧肿瘤细胞区域,利用该低氧环境,可以选择性地激活前体药物,使其分解产生AGT抑制剂,提高细胞对药物的敏感性。
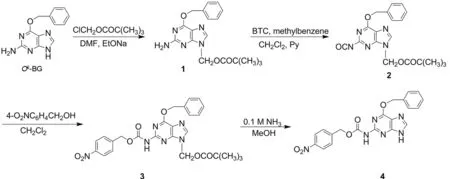
Scheme 1
为合成兼具良好AGT抑制活性和靶向性的新型抗肿瘤药物,本文以O6-BG为原料,与特戊酸氯甲酯经烷基化反应后,与三光气发生羰基化反应制得中间体(2); 2与对硝基苯甲醇发生加聚反应后,再经脱保护基反应合成了一个新型的O6-苄基鸟嘌呤衍生物——4-硝基苄基-[6-(苄氧基)-9H-嘌呤-2]氨基甲酸酯(4, Scheme 1),其结构经1H NMR和HR-ESI-MS表征。用CCK-8法研究了4对人脑神经胶质细胞(SF126, SF763和SF767)的细胞毒性。
1实验部分
1.1仪器与试剂
Bruker AC-P 400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Bruker Vertex 70型傅立叶变换红外光谱仪(KBr压片);TSQ Quantum型高效液相色谱-质谱联用仪。
O6-BG,纯度98%,百灵威科技有限公司;三光气(BTC),纯度99%,Sigma公司;其余所用试剂均为分析纯或优级纯。
1.2合成
(1) 1的合成
在反应瓶中加入O6-BG 0.24 g(1 mmol)和1 mol·L-1乙醇钠68 mg的乙醇(1 mL)溶液,超声10 min使其溶解;真空抽干乙醇,残余物用DMF(2 mL)溶解,搅拌下滴加特戊酸氯甲酯0.14 mL(1 mmol),滴毕,于室温反应30 min。真空抽干DMF,残余物经硅胶柱色谱[洗脱剂:V(石油醚) ∶V(乙酸乙酯)=1 ∶1]纯化得白色粉末1 0.26 g,产率72.3%, m.p.161~162 ℃;1H NMRδ: 1.09(s, 9H, CH3), 5.48 (s, 2H, CH2N), 5.96(s, 2H, CH2in Bn), 6.64(s, 2H, NH2), 7.34(m, 1H, ArH), 7.39(m, 2H, ArH), 7.50(m, 2H, ArH), 7.92(s, 1H, 8-H); ESI-MSm/z: 356{[M+H]+}。
(2) 2的合成
避光、氮气保护下,在圆底烧瓶中依次加入1 106 mg(0.3 mmol),无水二氯甲烷7 mL和无水吡啶0.5 mL,充分搅拌得溶液A。
冰浴冷却,在反应瓶中加入20%BTC 0.26 g(0.88 mmol)的甲苯(1.5 mL)溶液,搅拌下滴加溶液A,滴毕,反应20 h。升至室温得白色结晶2,直接投入下步反应。
(3) 3的合成
超声至2溶解,搅拌下加入对硝基苯甲醇0.11 g(0.7 mmol)的二氯甲烷(3 mL)溶液,反应2 h。反应液经硅胶柱层析[洗脱剂:V(二氯甲烷) ∶V(乙醇)=30 ∶1]纯化得白色粉末3 0.142 g,产率89%, m.p.146~148 ℃;1H NMRδ: 10.20(s, D2O exchangeable, 1H), 8.24(d,J=8.5 Hz, 2H ), 8.03(s, 1H), 7.33~7.61(m, 7H), 6.08(s, 2H), 5.62(s, 2H), 5.37(s, 2H), 1.16(s, 9H); ESI-MSm/z: 535{[M+H]+}。
(4) 4的合成
在圆底烧瓶中加入3 0.53 g(1 mmol)和0.1 mol·L-1氨的甲醇(40 mL)溶液,搅拌使其溶解;于室温反应约40 h(TLC跟踪)。析出白色粉末,水浴反应1 h。过滤,滤饼依次用甲醇和乙醚洗涤,干燥得白色粉末4 0.36 g,产率85%, m.p.233~235 ℃;1H NMRδ: 13.24(s, 1H), 10.51(s, 1H), 8.29~8.21(m, 3H), 7.74(d,J=8.7 Hz, 2H), 7.61~7.49(m, 2H), 7.46~7.26(m, 3H), 5.59(s, 2H), 5.34(s, 2H);13C NMRδ: 151.8, 151.7, 147.0, 144.7, 136.3, 128.9, 128.5, 128.3, 123.6, 67.5, 64.5; HR-ESI-MSm/z: Calcd for C20H16N6O5{[M+H]+}421.125 5, found 421.125 7。
1.3细胞毒性测定
以O6-BG为阳性对照,4和O6-BG作前体药物与CENUs抗肿瘤药物尼莫司汀(ACNU)协同作用于人脑神经胶质瘤细胞株SF126, SF763和SF767,采用CCK-8法测定4与ACNU联合用药的体外抗癌活性。
用完全培养基调整细胞浓度至5~7×104个/mL,接种于96孔板,每孔100 μL,培养过夜。依次用不同浓度的4的培养基处理细胞,同时设不含药物的阴性对照组和试剂空白对照组。每个剂量组设3组平行,分别于37°C, 5%CO2培养箱中和厌氧培养罐中培养24 h。更换培养基,每孔加入含10%CCK-8试剂的100 μL培养基,继续培养2~3 h。用酶标仪测定待测液在450 nm处的吸光值A450(A与活细胞数成正比,取平均值),计算细胞存活率[细胞存活率/%=(OD实验-OD空白)/OD对照-OD空白×100%],再计算IC50。
2结果与讨论
2.1合成
合成3时,反应仪器和试剂均需保证严格无水,否则可能导致合成失败。合成4时,需在氮气保护下,用注射器向反应母液中逐滴加入对硝基苯甲醇,使反应物充分反应。
2.2细胞毒性
表1为4的细胞毒性。由表1可见,在有氧条件下,4与ACNU的协同作用对SF126, SF763和SF767三种肿瘤细胞均无明显抑制效果,其IC50分别为0.8 mM, 1.2 mM和0.6 mM,与ACNU(0.85 mM, >1.2 mM, 0.9 mM) 无显著性差异;在无氧条件下,4与ACNU的协同作用对SF126, SF763和SF767三种肿瘤细胞具有显著的抑制效果,且明显优于ACNU单独作用和ACNU与O6-BG的协同作用效果。说明4可以低氧激活靶向作用于肿瘤细胞。
4作为一种低氧激活前体药物,其本身对细胞无毒或毒性很低,但在低氧条件下可被相应还原酶激活,释放具有细胞毒性的药物,从而发挥治疗作用,且对还原酶表达水平较低的正常组织影响较小,因此不论是抗肿瘤作用还是安全性均比传统化疗药物更具优势[14]。此外,由于4的结构中含有对硝基苄基,使其也能在低氧条件下靶向作用于实体瘤组织。研究表明,硝基芳烃化合物在低氧环境下很容易失去一个电子生成硝基阴离子自由基,而在有氧条件下与氧分子发生氧化反应迅速生成硝基芳烃或超氧歧化物[15]。正是利用硝基芳香化合物的这一特点,4的靶向作用性得以进一步增强。
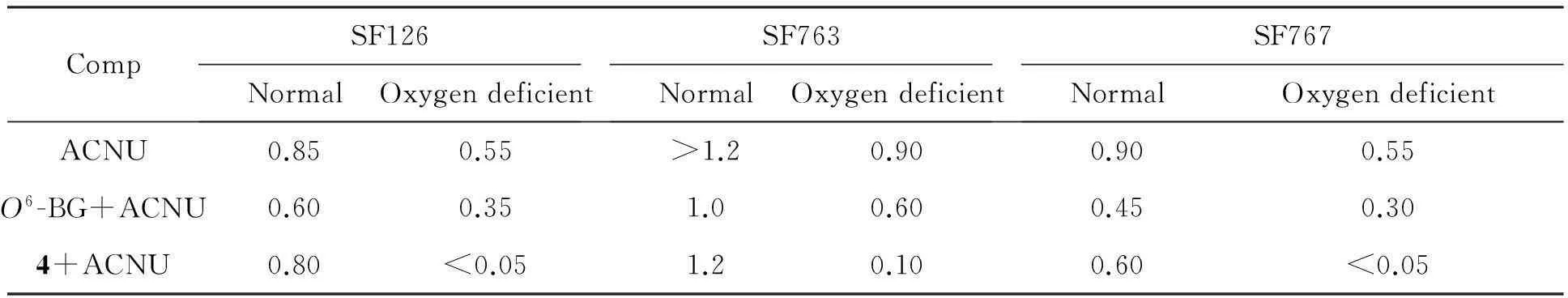
表1 4与ACNU联合用药的IC50
3结论
合成了低氧激活的4-硝基苄基-(6-(苄氧基)-9H-嘌呤-2)氨基甲酸酯(4)。用CCK-8法研究了4的细胞毒性。结果表明:在低氧条件下,4与ACUN的协同作用对人脑神经胶质瘤细胞SF126, SF763和SF767均具有显著的抑制作用,在有氧条件下无明显抑制效果,即4具有低氧激活靶向作用。低氧条件下,4作为前体药物可以显著抑制AGT活性,其对SF126, SF763和SF767三种肿瘤细胞的 IC50分别为0.04 mM, 0.1 mM和0.03 mM,均明显优于阳性对照药O6-BG。
参考文献
[1]Altiok N, Ersoz M, Koyuturk M. Estradiol induces JNK-dependent apoptosis in glioblastomacells [J].Oncol Lett,2011,2(6):1281-1285.
[2]白宝清,赵丽娇,钟儒刚. 亚硝基脲对DNA损伤作用的研究进展[J].化学通报,2010,3:212-219.
[3]吕申. 细胞DNA修复系统与肿瘤发生及化疗耐药性[J].大连医科大学学报,2012,34(6):519-524.
[4]Pegg A E. Repair ofO6-alkylguanine by alkyltransferases [J].Mutat Res,2000,462(2-3):83-100.
[5]Quinn J A, Pluda J, Dolan M E,etal. Phase II trial of carmustine plusO6-benzylguanine for patients with nitrosourea-resistant recurrent or progressive malignant glioma [J].J Clin Oncol,2002,20(9):2277-2283.
[6]Pegg A E, Dolan M E, Moschel R C. Structure,function,and inhibition ofO6-alkylguanine-DNA alkyltransferase[J].Prog Nucleic Acid Res Mol Biol,1995,51:167-223.
[7]Daniels D S, Tainer J A. Conserved structural motifs governing the stoichiometric repair of alkylated DNA byO6-alkylguanine-DNA alkyltransferase[J].Mutat Res,2000,460(3-4):151-163.
[8]Schilsky R L, Dolan M E, Bertucci D,etal. Phase I clinical and pharmacological study ofO6-benzylguanine followed by carmustine in patients with advanced cancer [J].Clin Cancer Res,2000,6(8):3025-3031.
[9]Sartorelli A C. Therapeutic attack of hypoxic cells of solid tumors:Presidential address[J].Cancer Res,1988,48(4):775-778.
[10]Chae M Y, Swenn K, Kanugula S,etal. 8-SubstitutedO6-benzylguanine,substituted 6(4)-(benzyloxy)pyrimidine,and related derivatives as inactivators of humanO6-alkylguanine-DNA alkyltransferase[J].J Med Chem,1995,38(2):359-365.
[11]Pegg A E. MammalianO6-alkylguanine-DNA alkyltransferase:Regulation and importance in response to alkylating carcinogenic and therapeutic agents[J].Cancer Res,1990,50(19):6119-6129.
[12]Margison G P, Povey A C, Kaina B,etal. Variability and regulation ofO6-alkylguanine-DNA alkyltransferase[J].Carcinogenesis,2003,24(4):625-635.
[13]Chae M Y, McDougall M G, Dolan M E,etal. SubstitutedO6-benzylguanine derivatives and their inactivation of humanO6-alkylguanine-DNA alkyltransferase[J].J Med Chem,1994,37(3):342-347.
[14]余长顺,欧阳洪贵,胡斌,等. 低氧激活的抗肿瘤药物及其研究近况[J].药学进展, 2012,36(2):65-72.
[15]Penketh P G, Shyam K, Baumann R P,etal. A Strategy for selectiveO6-alkylguanine-DNA alkyltransferase depletion under hypoxic conditions[J].Chem Biol Drus Des,2010,80(2):279-290.
《合成化学》变更期刊启事
为缩短出版周期,提高刊物的时效性,满足广大科研工作者发表科研成果、加快学术交流的需要,更好地为作者和读者服务,经四川省新闻出版局批准,《合成化学》从2015年1月起由原来的双月刊更改为月刊,每月20日出版。
欢迎广大作者踊跃投稿。
《合成化学》编辑部
Synthesis and Cytotoxicity of A Novel
O6-benzylguanine Derivative
LI Si-si1*,PENG Rui-zeng2
(1. Institute of Environmental Health and Related Product Safety, Chinese Center for Disease Control and Prevention,
Beijing 100021, China; 2. College of Life Science & Bioengineering, Beijing University of Technology, Beijing 100124, China)
Abstract:A novelO6-benzylguanine(O6-BG) derivative, 4-nitrobenzyl-[6-(benzyloxy)-9H-purin-2-yl]carbamate(4), was synthesized by a four-step reaction, usingO6-BG as starting material. The structure was characterized by1H NMR and HR-ESI-MS. The cytotoxicities of 4 were investigated by CCK-8 method. The results indicated that 4 exhibited better inhibition activities against human glioma cell lines(SF126, SF763 and SF767) with ACUN thanO6-BG under hypoxia conditions. IC50of 4 against SF126, SF763 and SF767 were 0.04 mM, 0.1 mM and 0.03 mM, respectively.
Keywords:O6-benzylguanine; hypoxia activated; 4-nitrobenzyl-[6-(benzyloxy)-9H-purin-2-yl]carbamate; synthesis; cytotoxicity
作者简介:李思思(1990-),女,汉族,北京人,硕士研究生,主要从事抗癌药物的研究。E-mail: lisisi918731@aliyun.com
收稿日期:2015-03-16;
修订日期:2015-12-03
中图分类号:0621.3
文献标志码:A
DOI:10.15952/j.cnki.cjsc.1005-1511.2016.02.15080
